



- Biochemia
- Biofizyka
- Biologia
- Biologia molekularna
- Biotechnologia
- Chemia
- Chemia analityczna
- Chemia nieorganiczna
- Chemia fizyczna
- Chemia organiczna
- Diagnostyka medyczna
- Ekologia
- Farmakologia
- Fizyka
- Inżynieria środowiskowa
- Medycyna
- Mikrobiologia
- Technologia chemiczna
- Zarządzanie projektami
- Badania kliniczne i przedkliniczne




Wybrane metody izolacji DNA
Odkrycie kwasów nukleinowych, zarówno DNA jak i RNA było kamieniem milowym dla nauki. Od czasu przedstawienia modelu DNA przez Watsona i Cricka w 1953 roku, zaczęto coraz dokładniej analizować kwasy nukleinowe, by w pełni poznać ich strukturę i funkcję. Odkrycie Watsona i Cricka zostało uznane za jedno z najznakomitszych w historii biologii [5].
Pomimo, iż istnieje wiele różnych metod izolacji kwasów nukleinowych, ciągle opracowywane są nowe procedury. Ich celem jest dostarczenie DNA lub RNA w jak największej ilości, a także najlepszej czystości i jakości. Nowe metody izolacji mają być efektywne, szybkie, wydajne i co najważniejsze tanie, tak by można było wykonywać wiele analiz na raz, nie zwiększając tym samym kosztów. Jedną z najbardziej uznanych metod izolacji jest ta, w której wykorzystuje się mieszaninę związków fenol: chloroform. Przez wielu metoda ta uznawana jest za najefektywniejszą [4], [2].
Jednym z najważniejszych etapów pracy z DNA stanowi prawidłowe pobranie materiału biologicznego, a także jego izolacja i przechowywanie. Bardzo ważny jest etap izolacji, ponieważ musi zapewnić on wysoką jakość preparatu DNA, dzięki czemu umożliwia to wykonanie dalszych analiz [4]. Wśród materiałów biologicznych najczęściej wykorzystywanych do izolacji DNA należy krew obwodowa (w przypadku człowieka i zwierząt), zaś w przypadku roślin są to liście i młode siewki. Ponadto w badaniach dotyczących człowieka wykorzystuje się także DNA izolowane z :
Obecnie metody izolacji można podzielić na 3 główne grupy:
1) Izolacja DNA z zastosowaniem mieszaniny fenol: chloroform( stosowane do odbiałczania preparatów)
2) Izolacja DNA, która przebiega z wysoleniem białek z otrzymanych lizatów komórek
3) Izolacja DNA z wykorzystaniem odpowiedniego nośnika, do którego DNA będzie się wiązać, następnie odmycie zanieczyszczeń i uwolnienie związanego DNA [4].
Według R.Słomskiego, autora książki Analiza DNA, pomimo wielu różnych metod izolacji, nadal jedną z najlepszych pozostaje izolacja z zastosowaniem mieszaniny fenolu i chloroformu. Metoda ta dostarcza preparaty, które charakteryzuje wysoki stopień czystości. W związku z tym, preparaty te można dalej z powodzeniem stosować np. do analizy restrykcyjnej, klonowania czy reakcji PCR [4].
Izolacja DNA z pełnej krwi obwodowej (Słomski i wsp.)
W badaniach dotyczących człowieka, jednym z najlepszych materiałów biologicznych do izolacji DNA jest pełna krew obwodowa. Do wykonania pełnych badań wystarcza już 1ml krwi. Na początkowym etapie izolacji DNA z krwi obwodowej lizie ulegają głównie tylko te komórki, które nie zawierają jądra komórkowego. Są one odrzucane razem z surowicą. Leukocyty są zbierane, Następnie przemywane i na końcu lizowane. Odbiałczanie preparatu przebiega przez zastosowanie ekstrakcji fenolem i chloroformem, bądź białka wytrąca się stosując np. NaCl lub LiCl. Stosując ekstrakcję fenolową bardzo ważne jest, aby pH było wyższe niż 7,8, a to ze względu na to, że przy niższym pH znaczna część wyizolowanego DNA pozostaje w fazie organicznej. W końcowym etapie procedury, otrzymane DNA wytrącane jest alkoholem etylowym lub izopropylowym [4].
Izolacja DNA z zastosowaniem ekstrakcji fenolem i chloroformem( z pełnej krwi obwodowej)
Na początkowym etapie izolacji należy przeprowadzić lizę komórek. W tym celu należy dodać 5 objętości buforu zawierającego NH4Cl (tj. 155 mM NH4Cl, 10 mM KHCO3, 0.1 mM Tris-HCl o pH=7.4). Próbkę krótko inkubować (ok. 5 min). Lizat zwirować przy 900xg przez 10 min (w celu uzyskania osadu jąder komórkowych). Otrzymane jądra komórkowe przemyć buforem PBS, i zawiesić w 2 ml buforu TE. Następnie należy ekstrahować DNA z fazy wodnej, w tym celu dodać równą objętość fenolu wysyconego buforem TE. Łagodnie wytrząsać próbkę przez 10 min. Rozdzielić otrzymane fazy na drodze wirowania : 2000xg, 5 min, Po wirowaniu zebrać fazę wodną i ponownie ekstrahować równą objętością mieszaniny: fenol: chloroform: alkohol izoamylowy w stosunku: 50:49:1. Zwirować, zebrać fazę wodną, dodać 0.1 objętości 3M octanu sodu i wytrącać DNA przez dodanie 2,5 objętości etanolu. Wytrącony osad DNA przemyć 75% etanolem i wysuszyć w temperaturze pokojowej lub w 37˚C. Po suszeniu DNA zawiesić w 500 μl buforu TE. Aby DNA całkowicie się rozpuściło, należy tak przygotowaną próbę pozostawić na ok. 24h (od wytrącenia DNA) [4].
Izolacja DNA z odsalaniem białek
10 ml krwi obwodowej należy pobrać do probówek zawierających 100 μl 10% EDTA (stężenie końcowe 0.1%). Do próbki dodać 30 ml buforu do lizy (tj. 155 mM NH4Cl, 10 mM KHCO3, 0.1 mM Tris-HCl o pH=7.4). Próbkę inkubować w temperaturze 0˚C prze 0.5h, kilkakrotnie mieszając. Lizat krwi zwirować w wirówce : 3000 rpm, 4˚C,10 min. Osad limfocytów ponownie zawiesić w buforze do lizy i zwirować (czynność tę powtórzyć trzykrotnie). Po wirowaniu, do osadu dodać 5 ml buforu SE (tj. 75 mM NaCl , 1 mM Na2EDTA o pH=8.0), 25 μl proteinazy K (10 mg/ml) oraz 250 μl SDS(20%). Całość dokładnie wymieszać i inkubować 16h w 55˚C. Po inkubacji uzyskany lizat intensywnie wymieszać z 750 μl 6M NaCl przez 15 sekund, Następnie dwukrotnie zwirować: 3600 rpm, 15 minut w temperaturze pokojowej. Otrzymane DNA wytrącić dwoma objętościami absolutnego etanolu, dalej przemyć 75% etanolem i rozpuścić w sterylnej H2O [4].
Izolacja DNA z zastosowaniem mocznika
10 ml krwi obwodowej pobrać do probówek zawierających EDTA (końcowe stężenie 0.1%), zamrozić na 24h w temp. -20˚C. Następnie krew zwirować: 3000rpm, 10 min, 4˚C. Otrzymany osad komórek zawiesić w roztworze do lizy (tj. 0,8% Triton X-100, 150 mM NaCl), ponownie odwirować: 12000-15000 rpm,10 min, 4˚C. Zlizować jądra komórkowe, zawieszając osad w 2 ml 7M mocznika, 0,5 ml 10% SDS, dodać 0,3 ml 10xstężonego buforu do lizy (tj. 3,5M NaCl, 0,013 M EDTA, 0,09M Tris-HCl, pH=8.0. Dokładnie wymieszać i dodać 7 ml tego samego buforu, tylko 1x stężonego(rozcieńczyć). Tak uzyskany lizat 2x ekstrahować równą objętością mieszaniny fenolu (wysyconego 0,1 M Tris-HCl, pH=8.0 ) i chloroformem (w stosunku 1:1). Za pomocą wirowania rozdzielić fazę wodną i fenolową (8000-10000 rpm, 10 min, 4˚C). Otrzymane DNA wytrącić przez dodanie 2 objętości absolutnego etanolu w obecności 0,3M octanu sodu . Osad DNA dwukrotnie przepłukać 1 ml 75% etanolu. Następnie rozpuścić DNA w 200 μl sterylnej wody. Przechowywać w 4˚C. Po wykonaniu dalszych badań DNA przechowywać w temp. -20˚C [4].
Izolacja DNA z zastosowaniem izotiocyjanianu guanidyny (GTC)
5 ml Krwi obwodowej pobrać na 50μl 10% EDTA (stężenie końcowe 0,1%). Do krwi dodać 25 ml buforu do lizy (tj. 155 mM NH4Cl, 10 mM KHCO3, 0,1 mM EDTA, pH=7.4). Całość wymieszać i inkubować 30 min w temp. 0˚C. Po inkubacji próbkę zwirować: 2500 rpm, 15 min, 4˚C. Supernatant odrzucić. Otrzymany osad zawiesić w 500 μl buforu do lizy, dokładnie wymieszać, a następnie inkubować w 0˚C przez 10 min. Po inkubacji dodać 2 ml GTC (tj. 4M izotiocyjanian guanidyny, 34 mM cytrynian sodu, 0,5% sarkosyl, 0,78% 2-merkaptoetanol, pH=7.0). Całość dokładnie wymieszać przez pipetowanie , inkubować 15 min w 0˚C. Następnie dodać 2,5 ml mieszaniny fenol: chloroform: alkohol izoamylowy, w stosunku 25:24:1, pH=8.0, dokładnie wymieszać. Próbkę zwirować: 3000 rpm, 15 min, 4˚C. Po wirowaniu zebrać supernatant. Dodać 2,5 ml chloroformu i znów dobrze wymieszać. Zwirować jak wcześniej, zebrać supernatant. Dodać 5 ml zimnego etanolu (-20˚C), lub 2 ml izopropanolu. Zebrać wytrącony osad i przemyć go 1 ml 75% etanolu. Przemyty osad suszyć w temperaturze pokojowej, lub w 37˚C. Na koniec wysuszony osad DNA rozpuścić w 300μl sterylnej wody [4].
Izolacja materiału genetycznego w celu identyfikacji osób zmarłych, w szczególności w sprawach karnych, bądź w wyniku klęsk żywiołowych, jest bardzo ciężka ze względu na materiał biologiczny, który często jest bardzo zdegradowany, z którego potem ma się izolować DNA.
Często materiał biologiczny pochodzi z rozkładającego się ciała, a do izolacji przeważnie wykorzystuje się albo mocno zdegradowane tkanki, albo kości, zęby lub paznokcie. Pracując na takim materiale należy brać pod uwagę wybór odpowiedniej metody izolacji [3].
Wybierając materiał do izolacji należy brać pod uwagę stopień jego rozkładu, czyli m.in. „marmurkowate” przebarwienia i pęcherze na skórze, obrzęki, zakażenia lub gnicie organów. Per Hoff-Olsen i wsp. przedstawili kilka różnych metod ekstrakcji DNA z rozłożonych tkanek ludzkich. Jako materiał biologiczny w swoich doświadczeniach wykorzystali fragmenty wątroby o wielkości 2,0x2,0x3,0 cm, pobrane za pomocą biopsji. Skrawki od razu zamrożono w -20˚C (do dalszej analizy i ekstrakcji) [3].
Ekstrakcja DNA metodą fenolowo chloroformową(zmodyfikowana po Sambrook i wsp.)
Do ok. 1ml próbki (w tym przypadku wątroby) dodać 5ml buforu do ekstrakcji pH=8.0 (10 mM Tris-HCl, 10mM EDTA, 100mM NaCl, 2% SDS). Łagodnie wymieszać przez 30 min w 37˚C. Następnie, dodać 50 ml proteinazy K(20 mg/ml) i delikatnie mieszać przez noc w 37˚C. Po nocnym mieszaniu, do próbki dodać 10 ml mieszaniny: chloroform: fenol : alkohol izoamylowy w stosunku : 49.5:49.5:1.0. Próbkę zwirować przy 3500 rpm przez 10 min. Po wirowaniu, górną warstwę przenieść do nowej probówki i wymieszać z 5ml mieszaniny chloroform: fenol: alkohol izoamylowy, po czym znów zwirować j.w. Górną warstwę próbki ponownie przenieść do nowej probówki i dodać 5ml mieszaniny chloroform: alkohol izoamylowy w stosunku 24:1 przed trzecim wirowaniem przy 3500 rpm (10 min). Po wirowaniu supernatantu przenieść do nowej probówki. Do otrzymanego nierozpuszczalnego DNA dodać 2-3 objętości zimnego, absolutnego alkoholu etylowego(do supernatantu). Roztwór odwirować przy maksymalnej prędkości przez 30 min. DNA przemyć przez wirowanie z maksymalną prędkością w 70% alkoholu, w niskiej temperaturze. Osad wysuszyć- przed rozpuszczeniem w 50 ml buforu TE o pH=7,6 (10mM Tris, 1mM EDTA). Następnie, roztwór wytrząsać przez noc [3].
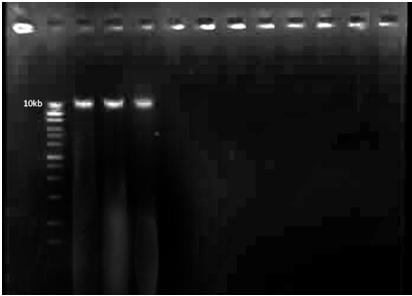
Zdjęcie: DNA genomowe (źródło: L. Koperwas).
Pomimo, iż istnieje wiele różnych metod izolacji kwasów nukleinowych, ciągle opracowywane są nowe procedury. Ich celem jest dostarczenie DNA lub RNA w jak największej ilości, a także najlepszej czystości i jakości. Nowe metody izolacji mają być efektywne, szybkie, wydajne i co najważniejsze tanie, tak by można było wykonywać wiele analiz na raz, nie zwiększając tym samym kosztów. Jedną z najbardziej uznanych metod izolacji jest ta, w której wykorzystuje się mieszaninę związków fenol: chloroform. Przez wielu metoda ta uznawana jest za najefektywniejszą [4], [2].
Jednym z najważniejszych etapów pracy z DNA stanowi prawidłowe pobranie materiału biologicznego, a także jego izolacja i przechowywanie. Bardzo ważny jest etap izolacji, ponieważ musi zapewnić on wysoką jakość preparatu DNA, dzięki czemu umożliwia to wykonanie dalszych analiz [4]. Wśród materiałów biologicznych najczęściej wykorzystywanych do izolacji DNA należy krew obwodowa (w przypadku człowieka i zwierząt), zaś w przypadku roślin są to liście i młode siewki. Ponadto w badaniach dotyczących człowieka wykorzystuje się także DNA izolowane z :
- -komórek nabłonka
- -hodowli fibroblastów
- -komórek płynu owodniowego (AFC), kosmówki (CVS)
- - komórek cebulek włosów.
Obecnie metody izolacji można podzielić na 3 główne grupy:
1) Izolacja DNA z zastosowaniem mieszaniny fenol: chloroform( stosowane do odbiałczania preparatów)
2) Izolacja DNA, która przebiega z wysoleniem białek z otrzymanych lizatów komórek
3) Izolacja DNA z wykorzystaniem odpowiedniego nośnika, do którego DNA będzie się wiązać, następnie odmycie zanieczyszczeń i uwolnienie związanego DNA [4].
Według R.Słomskiego, autora książki Analiza DNA, pomimo wielu różnych metod izolacji, nadal jedną z najlepszych pozostaje izolacja z zastosowaniem mieszaniny fenolu i chloroformu. Metoda ta dostarcza preparaty, które charakteryzuje wysoki stopień czystości. W związku z tym, preparaty te można dalej z powodzeniem stosować np. do analizy restrykcyjnej, klonowania czy reakcji PCR [4].
Izolacja DNA z pełnej krwi obwodowej (Słomski i wsp.)
W badaniach dotyczących człowieka, jednym z najlepszych materiałów biologicznych do izolacji DNA jest pełna krew obwodowa. Do wykonania pełnych badań wystarcza już 1ml krwi. Na początkowym etapie izolacji DNA z krwi obwodowej lizie ulegają głównie tylko te komórki, które nie zawierają jądra komórkowego. Są one odrzucane razem z surowicą. Leukocyty są zbierane, Następnie przemywane i na końcu lizowane. Odbiałczanie preparatu przebiega przez zastosowanie ekstrakcji fenolem i chloroformem, bądź białka wytrąca się stosując np. NaCl lub LiCl. Stosując ekstrakcję fenolową bardzo ważne jest, aby pH było wyższe niż 7,8, a to ze względu na to, że przy niższym pH znaczna część wyizolowanego DNA pozostaje w fazie organicznej. W końcowym etapie procedury, otrzymane DNA wytrącane jest alkoholem etylowym lub izopropylowym [4].
Izolacja DNA z zastosowaniem ekstrakcji fenolem i chloroformem( z pełnej krwi obwodowej)
Na początkowym etapie izolacji należy przeprowadzić lizę komórek. W tym celu należy dodać 5 objętości buforu zawierającego NH4Cl (tj. 155 mM NH4Cl, 10 mM KHCO3, 0.1 mM Tris-HCl o pH=7.4). Próbkę krótko inkubować (ok. 5 min). Lizat zwirować przy 900xg przez 10 min (w celu uzyskania osadu jąder komórkowych). Otrzymane jądra komórkowe przemyć buforem PBS, i zawiesić w 2 ml buforu TE. Następnie należy ekstrahować DNA z fazy wodnej, w tym celu dodać równą objętość fenolu wysyconego buforem TE. Łagodnie wytrząsać próbkę przez 10 min. Rozdzielić otrzymane fazy na drodze wirowania : 2000xg, 5 min, Po wirowaniu zebrać fazę wodną i ponownie ekstrahować równą objętością mieszaniny: fenol: chloroform: alkohol izoamylowy w stosunku: 50:49:1. Zwirować, zebrać fazę wodną, dodać 0.1 objętości 3M octanu sodu i wytrącać DNA przez dodanie 2,5 objętości etanolu. Wytrącony osad DNA przemyć 75% etanolem i wysuszyć w temperaturze pokojowej lub w 37˚C. Po suszeniu DNA zawiesić w 500 μl buforu TE. Aby DNA całkowicie się rozpuściło, należy tak przygotowaną próbę pozostawić na ok. 24h (od wytrącenia DNA) [4].
Izolacja DNA z odsalaniem białek
10 ml krwi obwodowej należy pobrać do probówek zawierających 100 μl 10% EDTA (stężenie końcowe 0.1%). Do próbki dodać 30 ml buforu do lizy (tj. 155 mM NH4Cl, 10 mM KHCO3, 0.1 mM Tris-HCl o pH=7.4). Próbkę inkubować w temperaturze 0˚C prze 0.5h, kilkakrotnie mieszając. Lizat krwi zwirować w wirówce : 3000 rpm, 4˚C,10 min. Osad limfocytów ponownie zawiesić w buforze do lizy i zwirować (czynność tę powtórzyć trzykrotnie). Po wirowaniu, do osadu dodać 5 ml buforu SE (tj. 75 mM NaCl , 1 mM Na2EDTA o pH=8.0), 25 μl proteinazy K (10 mg/ml) oraz 250 μl SDS(20%). Całość dokładnie wymieszać i inkubować 16h w 55˚C. Po inkubacji uzyskany lizat intensywnie wymieszać z 750 μl 6M NaCl przez 15 sekund, Następnie dwukrotnie zwirować: 3600 rpm, 15 minut w temperaturze pokojowej. Otrzymane DNA wytrącić dwoma objętościami absolutnego etanolu, dalej przemyć 75% etanolem i rozpuścić w sterylnej H2O [4].
Izolacja DNA z zastosowaniem mocznika
10 ml krwi obwodowej pobrać do probówek zawierających EDTA (końcowe stężenie 0.1%), zamrozić na 24h w temp. -20˚C. Następnie krew zwirować: 3000rpm, 10 min, 4˚C. Otrzymany osad komórek zawiesić w roztworze do lizy (tj. 0,8% Triton X-100, 150 mM NaCl), ponownie odwirować: 12000-15000 rpm,10 min, 4˚C. Zlizować jądra komórkowe, zawieszając osad w 2 ml 7M mocznika, 0,5 ml 10% SDS, dodać 0,3 ml 10xstężonego buforu do lizy (tj. 3,5M NaCl, 0,013 M EDTA, 0,09M Tris-HCl, pH=8.0. Dokładnie wymieszać i dodać 7 ml tego samego buforu, tylko 1x stężonego(rozcieńczyć). Tak uzyskany lizat 2x ekstrahować równą objętością mieszaniny fenolu (wysyconego 0,1 M Tris-HCl, pH=8.0 ) i chloroformem (w stosunku 1:1). Za pomocą wirowania rozdzielić fazę wodną i fenolową (8000-10000 rpm, 10 min, 4˚C). Otrzymane DNA wytrącić przez dodanie 2 objętości absolutnego etanolu w obecności 0,3M octanu sodu . Osad DNA dwukrotnie przepłukać 1 ml 75% etanolu. Następnie rozpuścić DNA w 200 μl sterylnej wody. Przechowywać w 4˚C. Po wykonaniu dalszych badań DNA przechowywać w temp. -20˚C [4].
Izolacja DNA z zastosowaniem izotiocyjanianu guanidyny (GTC)
5 ml Krwi obwodowej pobrać na 50μl 10% EDTA (stężenie końcowe 0,1%). Do krwi dodać 25 ml buforu do lizy (tj. 155 mM NH4Cl, 10 mM KHCO3, 0,1 mM EDTA, pH=7.4). Całość wymieszać i inkubować 30 min w temp. 0˚C. Po inkubacji próbkę zwirować: 2500 rpm, 15 min, 4˚C. Supernatant odrzucić. Otrzymany osad zawiesić w 500 μl buforu do lizy, dokładnie wymieszać, a następnie inkubować w 0˚C przez 10 min. Po inkubacji dodać 2 ml GTC (tj. 4M izotiocyjanian guanidyny, 34 mM cytrynian sodu, 0,5% sarkosyl, 0,78% 2-merkaptoetanol, pH=7.0). Całość dokładnie wymieszać przez pipetowanie , inkubować 15 min w 0˚C. Następnie dodać 2,5 ml mieszaniny fenol: chloroform: alkohol izoamylowy, w stosunku 25:24:1, pH=8.0, dokładnie wymieszać. Próbkę zwirować: 3000 rpm, 15 min, 4˚C. Po wirowaniu zebrać supernatant. Dodać 2,5 ml chloroformu i znów dobrze wymieszać. Zwirować jak wcześniej, zebrać supernatant. Dodać 5 ml zimnego etanolu (-20˚C), lub 2 ml izopropanolu. Zebrać wytrącony osad i przemyć go 1 ml 75% etanolu. Przemyty osad suszyć w temperaturze pokojowej, lub w 37˚C. Na koniec wysuszony osad DNA rozpuścić w 300μl sterylnej wody [4].
Izolacja materiału genetycznego w celu identyfikacji osób zmarłych, w szczególności w sprawach karnych, bądź w wyniku klęsk żywiołowych, jest bardzo ciężka ze względu na materiał biologiczny, który często jest bardzo zdegradowany, z którego potem ma się izolować DNA.
Często materiał biologiczny pochodzi z rozkładającego się ciała, a do izolacji przeważnie wykorzystuje się albo mocno zdegradowane tkanki, albo kości, zęby lub paznokcie. Pracując na takim materiale należy brać pod uwagę wybór odpowiedniej metody izolacji [3].
Wybierając materiał do izolacji należy brać pod uwagę stopień jego rozkładu, czyli m.in. „marmurkowate” przebarwienia i pęcherze na skórze, obrzęki, zakażenia lub gnicie organów. Per Hoff-Olsen i wsp. przedstawili kilka różnych metod ekstrakcji DNA z rozłożonych tkanek ludzkich. Jako materiał biologiczny w swoich doświadczeniach wykorzystali fragmenty wątroby o wielkości 2,0x2,0x3,0 cm, pobrane za pomocą biopsji. Skrawki od razu zamrożono w -20˚C (do dalszej analizy i ekstrakcji) [3].
Ekstrakcja DNA metodą fenolowo chloroformową(zmodyfikowana po Sambrook i wsp.)
Do ok. 1ml próbki (w tym przypadku wątroby) dodać 5ml buforu do ekstrakcji pH=8.0 (10 mM Tris-HCl, 10mM EDTA, 100mM NaCl, 2% SDS). Łagodnie wymieszać przez 30 min w 37˚C. Następnie, dodać 50 ml proteinazy K(20 mg/ml) i delikatnie mieszać przez noc w 37˚C. Po nocnym mieszaniu, do próbki dodać 10 ml mieszaniny: chloroform: fenol : alkohol izoamylowy w stosunku : 49.5:49.5:1.0. Próbkę zwirować przy 3500 rpm przez 10 min. Po wirowaniu, górną warstwę przenieść do nowej probówki i wymieszać z 5ml mieszaniny chloroform: fenol: alkohol izoamylowy, po czym znów zwirować j.w. Górną warstwę próbki ponownie przenieść do nowej probówki i dodać 5ml mieszaniny chloroform: alkohol izoamylowy w stosunku 24:1 przed trzecim wirowaniem przy 3500 rpm (10 min). Po wirowaniu supernatantu przenieść do nowej probówki. Do otrzymanego nierozpuszczalnego DNA dodać 2-3 objętości zimnego, absolutnego alkoholu etylowego(do supernatantu). Roztwór odwirować przy maksymalnej prędkości przez 30 min. DNA przemyć przez wirowanie z maksymalną prędkością w 70% alkoholu, w niskiej temperaturze. Osad wysuszyć- przed rozpuszczeniem w 50 ml buforu TE o pH=7,6 (10mM Tris, 1mM EDTA). Następnie, roztwór wytrząsać przez noc [3].
Zdjęcie: DNA genomowe (źródło: L. Koperwas).
Izolacja DNA z wykorzystaniem złoża Chelex (zmodyfikowana po Walsh i wsp.)
0,3-0,5 cm3 kawałek tkanki wątroby posiekać na drobno skalpelem, a następnie umieścić w 1,5ml probówce typu Eppendorf. Próbkę zmacerować (rozmiękczyć) w 50ml sterylnej wody za pomocą końcówki pipety. Następnie dodać : 150ml 20% Chelex-u, 2ml 10 mg/ml kinazy białkowej i 7 ml 1M DTT, całość dobrze wymieszać. Roztwór inkubować w 56˚C przez 30 min(ewentualnie w 37˚C przez noc). Po inkubacji należy przebić wieczko probówki Eppendorf za pomocą igły jednorazowego użytku, a następnie próbkę gotować przez 8 minut. Próbkę wirować przez 3 min w 13K. Supernatant przenieść do nowej probówki typu Eppendorf i płukać z wykorzystaniem 20% Chelex-u [3].
Mechanizm działania Chelex-u nie jest do końca poznany, jednak wiadomo, że jest on silnym chelatorem jonów metali, przez co zabezpiecza DNA przed rozpadem w wysokiej temperaturze (100˚C), a także chroni je przed degradacją przez nukleazy. Izolacja DNA metodą, która wykorzystuje Chelex, jest powszechnie znana i wykorzystywana w badaniach kryminalistycznych śladów DNA [6].
Ekstrakcja na filtrach z włókna szklanego(metoda ta stosowana była przez Jang’a i Lee)
Próbkę tkanki wątroby przemyć 1 ml buforu z proteinazą K (2,5 ml KCl, 750 μl 1M Tris-HCl o pH=8,3; 2,5 ml 10% IGEPAL CA-630 detergent (ICN Biomedical Inc.) + uzupełnić dH2O do 50 ml). Próbki, maksymalnie 0,3 cm3 umieścić na okrągłym filtrze z włókna szklanego o średnicy 0,7 cm (Schleicher i Schuell Inc, filtr klasy 50)i filtrze zawierającym tkankę nasączoną w 100 μl metanolu przez 10 minut. Metanol usunąć przez odessanie, a probówki z filtrem suszyć w 80˚C przez 15 minut. Jeżeli będzie to konieczne, wewnętrzne ścianki probówki osuszyć za pomocą wacika. Następnie, 120 μl buforu z proteinazą K z 500 μg/ml proteinazy K, dodać do próbki i całość inkubować w 55˚C przez 45 min. Proteinazę K zinaktywować przez gotowanie próbki w wodzie przez 10 min. Próbkę zwirować przez 10 sekund [3].
Metody izolacji DNA, w których wykorzystuje się czynniki chaotropowe takie jak m.in. NaI czy NaClO4, krzemionkę lub cząsteczki szkła, są powszechnie znane i bardzo często stosowane. GuSCN (rodanek guanidyny) często stosowany jest do oczyszczania i detekcji, zarówno DNA jak i RNA, ze względu na zdolność do przeprowadzenia lizy komórek, oraz inaktywację nukleaz. GuHCl (chlorowodorek guanidyny) również z powodzeniem może być stosowany do oczyszczania kwasów nukleinowych, jednakże jest on mniej skuteczny niż GuSCN w odniesieniu do hamowania RNaz [1].
Ekstrakcja DNA na krzemionce (ekstrakcja krzemionkowa, zmodyfikowana po Hőss i Pääbo)
W celu przygotowania zawiesiny krzemionki: 60g krzemionki +500 ml wody. Tak otrzymany roztwór zostawić na 24h w temperaturze pokojowej. Po tym czasie usunąć 430 ml supernatantu i ponownie dodać 500 ml wody. Następnie roztwór wstrząsnąć, w celu dystrybucji cząsteczek krzemionki. Wymieszany roztwór pozostawić na 5h w temperaturze pokojowej Na koniec, usunąć 440 ml supernatantu i ostatecznie dodać do roztworu 600 ml stężonego HCl.
2 ml buforu ekstrakcyjnego (tj. 10M izotiocyjanian guanidyny [GuSCN]; 0,1M Tris-HCl o pH=6,4;0,02M EDTA o pH=8.0, i 1,3% Triton-X) dodać do 1.0cm3 próbki wątroby przed rozpoczęciem inkubacji i łagodnego mieszania w 60˚C przez co najmniej 3 do 4 godzin, ewentualnie przez noc. Następnie roztwór wirować przy 6000rpm przez 5 minut. Po wirowaniu usunąć 600 μl z supernatantu i dodać 400μl buforu ekstrakcyjnego, oraz 40μl zawiesiny krzemionki [3].
W celu związania DNA do cząsteczek krzemionki roztwór pozostawić na 10-30 minut w temperaturze pokojowej, a następnie wirować przez 3 minuty przy prędkości maksymalnej przed usunięciem supernatantu. Aby uniknąć powstawania potencjalnie niebezpiecznych dla zdrowia produktów ubocznych, supernatant należy przechowywać w 10 M NaOH aż do bezpiecznego zniszczenia.
Osad krzemionkowy przemywać: dwa razy w 750μl buforu płuczącego o pH=6,4 (10M GuSCN; 0,1M Tris-HCl), dwa razy w 750μl 70% etanolu, oraz jeden raz w 750μl acetonu [3].
Jednym z rutynowych badań przeprowadzanym w zakładach medycyny sądowej jest określenie profili genetycznych zmarłych osób, o nieznanej tożsamości. Stopień zaawansowania rozkładu zwłok, determinuje wybór materiału biologicznego, z którego izoluje się DNA. Bardzo zaawansowany proces gnilny tkanek miękkich, bądź zwęglenie lub całkowite ze szkieletowanie ciała powoduje, że jedynym źródłem, z którego można wyizolować DNA są kości, zęby lub paznokcie ofiary [2].
Kości, zęby i paznokcie, zaliczane są do tkanek twardych. Są one odporne na procesy gnilne i autolizę(w zależności od warunków środowiskowych, w których się znajdują). Czynniki zewnętrzne takie jak : światło, wilgotność, wysoka lub niska temperatura, a także woda i suche powietrze, są czynnikami, które wpływają na jakość i ilość izolowanego DNA. Ponadto, różnorodne bakterie, grzyby i nekrofagi, uwalniają substancje organiczne, które są dodatkowymi czynnikami zanieczyszczającymi, a także degradującymi materiał genetyczny. Do ustalenia profili genetycznych możliwe jest wykorzystanie materiału biologicznego w postaci kości, zębów i paznokci, z których następnie izoluje się DNA [2].
Izolacja DNA z kości
Kości, których ma być izolowane DNA należy poddać obróbce wstępnej, a następnie spiłowaniu. Do badań wykorzystuje się proszek kostny. Izolacja DNA z takiego materiału musi przebiegać w bardzo rygorystycznych warunkach sterylności. W doświadczeniu, przeprowadzonym przez E. Kapińska i Z. Szczerkowską, wykorzystano 4 różne metody izolacji DNA z kości:
1) Klasyczną metodę fenolowo-chloroformową
2) Metodę octanową
3) Izolację DNA z zastosowaniem złoża krzemionkowego wg metody opisanej przez Hőssa i Pääbo, a także
4) Komercyjny System firmy Promega (DNA IQTM) [2].
Do każdej z wymienionych metod izolacji, w doświadczeniu użyto ok. 1 g sproszkowanych kości (kości jednego rodzaju, np. kość udowa). Różne metody izolacji pozwalają na uzyskanie różnych ilości wyizolowanego DNA. W przypadku izolacji DNA z kości, które narażone były na działanie wysokiej temperatury (pożar), najbardziej wydajną metodą izolacji w doświadczeniu przeprowadzonym przez E. Kapińską i Z. Szczerkowską, okazała się klasyczna metoda fenolowo-chloroformowa. W przeprowadzonym doświadczeniu (Kapińska i Szczerkowska) o połowę mniej materiału genetycznego uzyskano przy zastosowaniu komercyjnego zestawu DNA IQTM System (Promega) oraz metodą z wykorzystaniem złoża krzemionkowego. Stosując ekstrakcję octanowa nie udało się wyizolować odpowiedniej ilości DNA(izolowanego z kości), co sugeruje, że metoda ta jest nieefektywna w przypadku izolacji z takiego materiału biologicznego. Tak więc, w doświadczeniu przeprowadzonym przez Kopalińską i Szczerkowską najlepszą metodą z wyboru, okazała się izolacja fenolowo-chloroformowa, pomimo jej stosunkowo dużej toksyczności i czasochłonności [2], [3].
Literatura:
[1]. Boom R, Sol C.J, Salimans M.M, Jansen C.L, Wertheim—van Dillen P.M, van der Noordaa J, 1990. Rapid and simple method for purification of nucleic acids. JOURNAL OF CLINICAL MICROBIOLOGY, Mar. 1990, p. 495-503. Vol. 28, No. 3
[2]. Kapińska E, Szczerkowska Z, 2004. PERSONAL IDENTIFICATION BASED ON NUCLEAR DNA EXTRACTED FROM BONES OF DECEASED INDIVIDUALS. Problems of Forensic Sciences, vol. LX, 2004, 104–116
[3]. Hoff-Olsen P, Mevag B, StaalstrØm, Hovde B, Egeland T, Olaisen B, 1999. Extraction of DNA from decomposed human tissue. An evaluation of five extraction methods for short tandem repeat typing. Forensic Science International 105 (1999) 171 –183
[4]. Słomski R, 2008. Analiza DNA, teoria I praktyka. Wydawnictwo Uniwersytetu Przyrodniczego w Poznaniu, Poznań 2008. S.44-53
[5]. Stryer L, 2003. Biochemia. Przekład zbiorowy pod redakcją: Augustyniak J, Michejda J, z czwartego wydania amerykańskiego. Wydawnictwo Naukowe PWN, Warszawa 2003, s. 76-83, 91-92.
[6].http://www.e-biotechnologia.pl/Artykuly/Porownanie-wyciszenia ekspresji-genu/
Opracowała: Lidia Koperwas
0,3-0,5 cm3 kawałek tkanki wątroby posiekać na drobno skalpelem, a następnie umieścić w 1,5ml probówce typu Eppendorf. Próbkę zmacerować (rozmiękczyć) w 50ml sterylnej wody za pomocą końcówki pipety. Następnie dodać : 150ml 20% Chelex-u, 2ml 10 mg/ml kinazy białkowej i 7 ml 1M DTT, całość dobrze wymieszać. Roztwór inkubować w 56˚C przez 30 min(ewentualnie w 37˚C przez noc). Po inkubacji należy przebić wieczko probówki Eppendorf za pomocą igły jednorazowego użytku, a następnie próbkę gotować przez 8 minut. Próbkę wirować przez 3 min w 13K. Supernatant przenieść do nowej probówki typu Eppendorf i płukać z wykorzystaniem 20% Chelex-u [3].
Mechanizm działania Chelex-u nie jest do końca poznany, jednak wiadomo, że jest on silnym chelatorem jonów metali, przez co zabezpiecza DNA przed rozpadem w wysokiej temperaturze (100˚C), a także chroni je przed degradacją przez nukleazy. Izolacja DNA metodą, która wykorzystuje Chelex, jest powszechnie znana i wykorzystywana w badaniach kryminalistycznych śladów DNA [6].
Ekstrakcja na filtrach z włókna szklanego(metoda ta stosowana była przez Jang’a i Lee)
Próbkę tkanki wątroby przemyć 1 ml buforu z proteinazą K (2,5 ml KCl, 750 μl 1M Tris-HCl o pH=8,3; 2,5 ml 10% IGEPAL CA-630 detergent (ICN Biomedical Inc.) + uzupełnić dH2O do 50 ml). Próbki, maksymalnie 0,3 cm3 umieścić na okrągłym filtrze z włókna szklanego o średnicy 0,7 cm (Schleicher i Schuell Inc, filtr klasy 50)i filtrze zawierającym tkankę nasączoną w 100 μl metanolu przez 10 minut. Metanol usunąć przez odessanie, a probówki z filtrem suszyć w 80˚C przez 15 minut. Jeżeli będzie to konieczne, wewnętrzne ścianki probówki osuszyć za pomocą wacika. Następnie, 120 μl buforu z proteinazą K z 500 μg/ml proteinazy K, dodać do próbki i całość inkubować w 55˚C przez 45 min. Proteinazę K zinaktywować przez gotowanie próbki w wodzie przez 10 min. Próbkę zwirować przez 10 sekund [3].
Metody izolacji DNA, w których wykorzystuje się czynniki chaotropowe takie jak m.in. NaI czy NaClO4, krzemionkę lub cząsteczki szkła, są powszechnie znane i bardzo często stosowane. GuSCN (rodanek guanidyny) często stosowany jest do oczyszczania i detekcji, zarówno DNA jak i RNA, ze względu na zdolność do przeprowadzenia lizy komórek, oraz inaktywację nukleaz. GuHCl (chlorowodorek guanidyny) również z powodzeniem może być stosowany do oczyszczania kwasów nukleinowych, jednakże jest on mniej skuteczny niż GuSCN w odniesieniu do hamowania RNaz [1].
Ekstrakcja DNA na krzemionce (ekstrakcja krzemionkowa, zmodyfikowana po Hőss i Pääbo)
W celu przygotowania zawiesiny krzemionki: 60g krzemionki +500 ml wody. Tak otrzymany roztwór zostawić na 24h w temperaturze pokojowej. Po tym czasie usunąć 430 ml supernatantu i ponownie dodać 500 ml wody. Następnie roztwór wstrząsnąć, w celu dystrybucji cząsteczek krzemionki. Wymieszany roztwór pozostawić na 5h w temperaturze pokojowej Na koniec, usunąć 440 ml supernatantu i ostatecznie dodać do roztworu 600 ml stężonego HCl.
2 ml buforu ekstrakcyjnego (tj. 10M izotiocyjanian guanidyny [GuSCN]; 0,1M Tris-HCl o pH=6,4;0,02M EDTA o pH=8.0, i 1,3% Triton-X) dodać do 1.0cm3 próbki wątroby przed rozpoczęciem inkubacji i łagodnego mieszania w 60˚C przez co najmniej 3 do 4 godzin, ewentualnie przez noc. Następnie roztwór wirować przy 6000rpm przez 5 minut. Po wirowaniu usunąć 600 μl z supernatantu i dodać 400μl buforu ekstrakcyjnego, oraz 40μl zawiesiny krzemionki [3].
W celu związania DNA do cząsteczek krzemionki roztwór pozostawić na 10-30 minut w temperaturze pokojowej, a następnie wirować przez 3 minuty przy prędkości maksymalnej przed usunięciem supernatantu. Aby uniknąć powstawania potencjalnie niebezpiecznych dla zdrowia produktów ubocznych, supernatant należy przechowywać w 10 M NaOH aż do bezpiecznego zniszczenia.
Osad krzemionkowy przemywać: dwa razy w 750μl buforu płuczącego o pH=6,4 (10M GuSCN; 0,1M Tris-HCl), dwa razy w 750μl 70% etanolu, oraz jeden raz w 750μl acetonu [3].
Jednym z rutynowych badań przeprowadzanym w zakładach medycyny sądowej jest określenie profili genetycznych zmarłych osób, o nieznanej tożsamości. Stopień zaawansowania rozkładu zwłok, determinuje wybór materiału biologicznego, z którego izoluje się DNA. Bardzo zaawansowany proces gnilny tkanek miękkich, bądź zwęglenie lub całkowite ze szkieletowanie ciała powoduje, że jedynym źródłem, z którego można wyizolować DNA są kości, zęby lub paznokcie ofiary [2].
Kości, zęby i paznokcie, zaliczane są do tkanek twardych. Są one odporne na procesy gnilne i autolizę(w zależności od warunków środowiskowych, w których się znajdują). Czynniki zewnętrzne takie jak : światło, wilgotność, wysoka lub niska temperatura, a także woda i suche powietrze, są czynnikami, które wpływają na jakość i ilość izolowanego DNA. Ponadto, różnorodne bakterie, grzyby i nekrofagi, uwalniają substancje organiczne, które są dodatkowymi czynnikami zanieczyszczającymi, a także degradującymi materiał genetyczny. Do ustalenia profili genetycznych możliwe jest wykorzystanie materiału biologicznego w postaci kości, zębów i paznokci, z których następnie izoluje się DNA [2].
Izolacja DNA z kości
Kości, których ma być izolowane DNA należy poddać obróbce wstępnej, a następnie spiłowaniu. Do badań wykorzystuje się proszek kostny. Izolacja DNA z takiego materiału musi przebiegać w bardzo rygorystycznych warunkach sterylności. W doświadczeniu, przeprowadzonym przez E. Kapińska i Z. Szczerkowską, wykorzystano 4 różne metody izolacji DNA z kości:
1) Klasyczną metodę fenolowo-chloroformową
2) Metodę octanową
3) Izolację DNA z zastosowaniem złoża krzemionkowego wg metody opisanej przez Hőssa i Pääbo, a także
4) Komercyjny System firmy Promega (DNA IQTM) [2].
Do każdej z wymienionych metod izolacji, w doświadczeniu użyto ok. 1 g sproszkowanych kości (kości jednego rodzaju, np. kość udowa). Różne metody izolacji pozwalają na uzyskanie różnych ilości wyizolowanego DNA. W przypadku izolacji DNA z kości, które narażone były na działanie wysokiej temperatury (pożar), najbardziej wydajną metodą izolacji w doświadczeniu przeprowadzonym przez E. Kapińską i Z. Szczerkowską, okazała się klasyczna metoda fenolowo-chloroformowa. W przeprowadzonym doświadczeniu (Kapińska i Szczerkowska) o połowę mniej materiału genetycznego uzyskano przy zastosowaniu komercyjnego zestawu DNA IQTM System (Promega) oraz metodą z wykorzystaniem złoża krzemionkowego. Stosując ekstrakcję octanowa nie udało się wyizolować odpowiedniej ilości DNA(izolowanego z kości), co sugeruje, że metoda ta jest nieefektywna w przypadku izolacji z takiego materiału biologicznego. Tak więc, w doświadczeniu przeprowadzonym przez Kopalińską i Szczerkowską najlepszą metodą z wyboru, okazała się izolacja fenolowo-chloroformowa, pomimo jej stosunkowo dużej toksyczności i czasochłonności [2], [3].
Literatura:
[1]. Boom R, Sol C.J, Salimans M.M, Jansen C.L, Wertheim—van Dillen P.M, van der Noordaa J, 1990. Rapid and simple method for purification of nucleic acids. JOURNAL OF CLINICAL MICROBIOLOGY, Mar. 1990, p. 495-503. Vol. 28, No. 3
[2]. Kapińska E, Szczerkowska Z, 2004. PERSONAL IDENTIFICATION BASED ON NUCLEAR DNA EXTRACTED FROM BONES OF DECEASED INDIVIDUALS. Problems of Forensic Sciences, vol. LX, 2004, 104–116
[3]. Hoff-Olsen P, Mevag B, StaalstrØm, Hovde B, Egeland T, Olaisen B, 1999. Extraction of DNA from decomposed human tissue. An evaluation of five extraction methods for short tandem repeat typing. Forensic Science International 105 (1999) 171 –183
[4]. Słomski R, 2008. Analiza DNA, teoria I praktyka. Wydawnictwo Uniwersytetu Przyrodniczego w Poznaniu, Poznań 2008. S.44-53
[5]. Stryer L, 2003. Biochemia. Przekład zbiorowy pod redakcją: Augustyniak J, Michejda J, z czwartego wydania amerykańskiego. Wydawnictwo Naukowe PWN, Warszawa 2003, s. 76-83, 91-92.
[6].http://www.e-biotechnologia.pl/Artykuly/Porownanie-wyciszenia ekspresji-genu/
Opracowała: Lidia Koperwas
Recenzje