



- Biochemia
- Biofizyka
- Biologia
- Biologia molekularna
- Biotechnologia
- Chemia
- Chemia analityczna
- Chemia nieorganiczna
- Chemia fizyczna
- Chemia organiczna
- Diagnostyka medyczna
- Ekologia
- Farmakologia
- Fizyka
- Inżynieria środowiskowa
- Medycyna
- Mikrobiologia
- Technologia chemiczna
- Zarządzanie projektami
- Badania kliniczne i przedkliniczne




Terapeutyczne właściwości emiterów promieniowania beta minus
 Emiterami promieniowania beta minus są izotopy promieniotwórcze pierwiastków charakteryzujące się rozpadem, w którym emitowany jest elektron (promieniowanie beta minus, β-) oraz antyneutrino elektronowe. W wyniku takiej reakcji jądrowej liczba masowa pierwiastka pozostaje bez zmian, zaś liczba atomowa wzrasta o jeden, gdyż jeden z neutronów w jądrze atomowym rozpada się na elektron, proton oraz antyneutrino elektronowe. Rozpadowi beta minus często towarzyszy promieniowanie gamma, zaś dla niektórych jąder emisja protonów lub neutronów. Istotną właściwością promieniowania beta minus jest jego zasięg i energia. Energia takiego promieniowania jest większa od promieniowania gamma, a za razem mniejsza od promieniowania alfa. Natomiast zasięg jest większy od promieniowania alfa i mniejszy od gamma, dzięki czemu znalazło ono zastosowanie w aplikacja współczesnej medycyny nuklearnej, a zwłaszcza w terapii celowanej guzów nowotworowych.
Emiterami promieniowania beta minus są izotopy promieniotwórcze pierwiastków charakteryzujące się rozpadem, w którym emitowany jest elektron (promieniowanie beta minus, β-) oraz antyneutrino elektronowe. W wyniku takiej reakcji jądrowej liczba masowa pierwiastka pozostaje bez zmian, zaś liczba atomowa wzrasta o jeden, gdyż jeden z neutronów w jądrze atomowym rozpada się na elektron, proton oraz antyneutrino elektronowe. Rozpadowi beta minus często towarzyszy promieniowanie gamma, zaś dla niektórych jąder emisja protonów lub neutronów. Istotną właściwością promieniowania beta minus jest jego zasięg i energia. Energia takiego promieniowania jest większa od promieniowania gamma, a za razem mniejsza od promieniowania alfa. Natomiast zasięg jest większy od promieniowania alfa i mniejszy od gamma, dzięki czemu znalazło ono zastosowanie w aplikacja współczesnej medycyny nuklearnej, a zwłaszcza w terapii celowanej guzów nowotworowych.
Wprowadzenie
W medycynie nuklearnej do znakowania biomolekuł o przeznaczeniu terapeutycznym stosuje się obecnie emitery promieniowania β- (beta minus) takie jak miedź-67 (67Cu), itr-90 (90Y), jod-131 (131I), samar-153 (153Sm), lutet-177 (177Lu), ren-186 (186Re) oraz ren-188 (188Re). Dzięki niskiej energii emitowanego promieniowania miedź-67, jod-131 oraz lutet-177 są idealne do unicestwiania małych skupisk komórek nowotworowych. Ren-186 ze średnioenergetycznym promieniowaniem β-, o wiele lepiej nadaje się do niszczenia średniego rozmiaru skupisk komórek rakowych. Natomiast itr-90 oraz ren-188 emitujące promieniowanie o wysokiej energii są bardzo korzystne w leczeniu dużych złogów komórek nowotworowych. Rodzaj radionuklidu musi być starannie dobrany w celu uniknięcia wchłaniania większej części emitowanego promieniowania przez otaczające chorą tkankę zdrowe komórki.
Najbardziej znanym i czystym emiterem promieniowania beta minus jest itr-90 (t1/2=64 h). Ten otrzymywany generatorowo radionuklid jest dobrze poznanym radionuklidem terapeutycznym. Jony itru oraz lantanowców, takich jak lutet i samar występują na +3 stopniu utlenienia. Ze względu na duże rozmiary, liczby koordynacyjne tych jonów przyjmują wartości pomiędzy 7 i 10. Dzięki bardzo zbliżonym właściwościom fizycznym, do ich kompleksowania używane są zarówno pochodne acyklicznego DTPA jak i cyklicznego DOTA.
Pierwsze radiofarmaceutyki oparte na emiterach promieniowania beta minus
Prekursorem badań nad związkami peptydowymi znakowanymi izotopami do radioterapii celowanej guzów nowotworowych stała się SRIF, a raczej jej analog zwany oktreotydem [1]. Pierwszym zarejestrowanym radiofarmaceutykiem diagnostycznym opartym na oktreotydzie był OctreoScan ([111In-DTPA-D-Phe1]-oktreotyd) [2] (Rys. 1). Początkowo w badaniach nad oktreotydem skupiono się na radionuklidach takich jak 111In, 90Y oraz 68Ga, które wprowadzano do struktury analogów somatostatyny zwłaszcza za pośrednictwem DOTA [3].
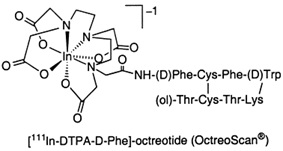
Rysunek 1. Struktura radiofarmaceutyku OctreoScan [4].
DOTA jako chelator 177Luznalazł zastosowanie w radiofarmaceutykach opartych na oktereotydzie w postaci [177Lu-DOTA-D-Phe1,Tyr3]-oktreotydu oraz [177Lu-DOTA-D-Phe1,Tyr3]-oktreotanu. Z czasem podjęto próby wykorzystania DTPA jako chelatora jonów lutetu-177.
Różnica w stosunku do wcześniej stosowanego chelatora DOTA polegała na tym, że proces znakowania z wykorzystaniem DTPA przeprowadzano w dużo niższej temperaturze. Dodatek chelatora DTPA rozwiązywał problem akumulacji niezwiązanego 177Lu w kościach pacjentów. Stwierdzono, że chelator DTPA wiąże uwolnione w ustroju jony 177Lu3+ i szybko ulega wydaleniu przez nerki, przez co zapobiega jego akumulacji w kościach [5].
Kompleks DTPA z 177Lu zastosowano w znakowaniu przeciwciał monoklonalnych (mAb), takich jak 2Rs15dHIS czy cG250 stosowanych w radioimmunoterapii raka nerki [6]. W badaniach porównawczych obok 177Lu przebadano izotopy 131I, 90Y oraz 186Re. Do związania radioizotopów ze strukturą mAb zastosowano chelatory takie jak cDTPA, SCN-Bz-DTPA oraz DOTA i stwierdzono, że izotop 177Lu wykazał najlepsze działanie przeciwnowotworowe. Prawdopodobnie było to wynikiem optymalnej charakterystyki promieniowania dla niewielkich guzów nowotworowych.
Znakowanie izotopami jodu molekuł biologicznie czynnych
Molekuły biologicznie czynne naturalnie występują w bardzo niskich stężeniach, których nie można analizować tradycyjnymi metodami. Wprowadzenie radioizotopu do struktury białka pozwala obniżyć próg detekcji przy zastosowaniu detektorów promieniowania beta minus lub gamma. Radioznacznikowanie izotopami jodu jest techniką polegającą na włączeniu w strukturę peptydu izotopów jodu-123 (123I), jodu-125 (125I) lub jodu-131 (131I), przede wszystkim w celu przeprowadzenia badań in vitro, ale izotopy te mogą też być wprowadzone w celach terapeutycznych lub diagnostycznych medycyny nuklearnej.
Obecnie istnieją trzy podstawowe metody jodowania, które są powszechnie wykorzystywane do znakowania biomolekuł używanych w terapii i diagnostyce medycznej. Mowa tu o metodzie z użyciem ampuł Iodo-Gen, metodzie wykorzystującej jako katalizatora chloraminę-T oraz metodzie z wykorzystaniem katalitycznych właściwości laktoperoksydazy. W zależności od stosunku molowego użytego jodu do grup tyrozynowych i histydynowych występujących w strukturze peptydu oraz od wydajności katalizatora, mogą powstawać pochodne monojodowe oraz pochodne jodowe wyższych rzędów. Wprowadzenie jodu do struktury białka lub peptydu z reguły nie wpływa na jego właściwości ze względu na niewielkie rozmiary atomu w porównaniu do całej biomolekuły.
Otrzymywanie radiofarmaceutyków peptydowych w roztworze i na podłożu stałym
Syntezę radiofarmaceutyku opartego na nośniku peptydowym można zrealizować na dwa sposoby. W pierwszym przypadku syntezę radiofarmaceutyku przeprowadza się czteroetapowo w roztworach organicznych, przy czym każdy etap zakończony jest wydzielaniem i ewentualnym oczyszczaniem produktu [7]. Wykorzystywane nośniki radionuklidów to najczęściej biologicznie czynne białka oraz antyciała monoklonalne. W drugim przypadku całą syntezę z poszczególnymi etapami protekcji i deprotekcji przeprowadza się na podłożu stałym i kończy znakowaniem otrzymanego koniugatu izotopem promieniotwórczym [8]. Proces takiej syntezy jest dwuetapowy.
Pierwszym etapem syntezy w roztworze jest odpowiednie zablokowanie, bądź przetworzenie grup funkcyjnych występujących na nośniku (wektorze) peptydowym w celu związania go z chelatorem (Rys. 2). Etap ten nazywany jest protekcją, natomiast kolejny - reakcją sprzęgania (koniugacji). Od chwili, gdy nośnik peptydowy połączony jest z chelatorem, otrzymany związek nazywany jest koniugatem.
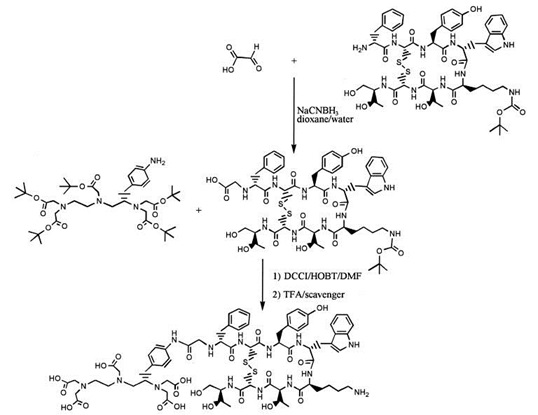
Rysunek 2. Etapy syntezy radiofarmaceutyku w roztworze DMF (dimetyloformamid) [7]. Protekcja, sprzęganie oraz deprotekcja.
Przy syntezie radiofarmaceutyków peptydowych najczęściej wykorzystywane są cykliczne chelatory dwufunkcyjne, będące pochodnymi DOTA, NOTA oraz TETA. Niektórymi acyklicznymi chelatorami wykorzystywanymi w tworzeniu koniugatów peptydowych są pochodne DTPA, liniowe fosfiny oraz aminotiole.
Przy zastosowaniu radioizotopu 99mTc bardzo popularnym chelatorem jest HYNIC w połączeniu z innymi liniowymi chelatorami dwufunkcyjnymi.
W celu połączenia nośnika z ligandem, na N- lub C-końcu nośnika utworzona zostaje grupa aktywna, która ułatwia utworzenie wiązania kowalencyjnego. W zależności od tego czy na końcu peptydu znajduje się grupa aminowa, hydroksylowa czy karboksylowa, stosowane są różne reagenty.
Trzeci etap syntezy w roztworze nosi nazwę deprotekcji. Na tym etapie odblokowywane są grupy funkcyjne koniugatu najczęściej za pomocą kwasu trifluorooctowego. Etap czwarty i ostatni, to znakowanie izotopem promieniotwórczym. Po efektywnym wyznakowaniu otrzymywany jest gotowy radiofarmaceutyk, który po wieloetapowej kontroli i testach może zostać podany pacjentowi. Wszystkie wymienione etapy syntezy radiofarmaceutyku peptydowego zakończone są oczyszczaniem produktu za pomocą technik chromatograficznych.
Synteza na podłożu stałym rozpoczyna się od wybrania odpowiedniego podłoża do rozbudowy struktury peptydu (Rys. 3). Wybór dokonywany jest w zależności od tego, który fragment nośnika odpowiedzialny jest za wiązanie do miejsca aktywnego receptora. Jeżeli jest to fragment na C-końcu nośnika, wówczas podłoże powinno być wysycone grupami karboksylowymi, hydroksylowymi lub innymi reagentami dla grupy aminowej znajdującej się po stronie przeciwnej.
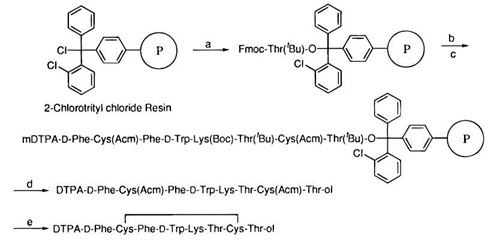
Rysunek 3. Etapy syntezy radiofarmaceutyku na podłożu stałym [8]. Wybranie podłoża P (a), wydłużanie łańcucha - elongacja (b,c), deprotekcja (d), utworzenie mostka siarczkowego (e). Związek gotowy do radioznacznikowania.
C-koniec nośnika w tej sytuacji powinien zostać odpowiednio zablokowany. W sytuacji odwrotnej, gdy aktywny jest N-koniec peptydu, wówczas podłoże powinno zawierać grupy aminowe lub inne reagenty dla grupy karboksylowej, bądź hydroksylowej zajmującej C-koniec peptydu, zaś N-koniec powinien podlegać protekcji.Wprowadzanie kolejnych aminokwasów do struktury peptydu podczas syntezy na podłożu stałym nazywane jest wydłużaniem łańcucha (elongacja).
Rozbudowana struktura peptydu posiada unikatową sekwencję, do której przyłączony zostaje chelator, a następnie cały koniugat zostaje poddany deprotekcji skutkującej dodatkowo odcięciem od podłoża stałego. Etap końcowy syntezy radiofarmaceutyku zarówno na podłożu stałym, jak i w roztworze to radioznacznikowanie izotopem pierwiastka emitującego promieniowanie beta minus.
Podsumowanie
Terapeutyczne właściwości radiofarmaceutyków będących emiterami promieniowania beta minus dotychczas objawiły się w zwalczaniu guzów nowotworowych, takich jak gruczolakoraków (wydzielających ACTH, PRL, TSH, GH), raku trzustki, rakach centralnego układu nerwowego (rdzeniaki, nerwiaki, oponiaki), rakach peryferycznego układu nerwowego (przyzwojaki), raku prostaty, piersi, płuc oraz okrężnicy. Typowymi nośnikami izotopów emitujących promieniowanie beta minus są peptydowe analogi somatostatyny typu oktreotyd. Z roku na rok pojawiają się nowe nośniki peptydowe, jak na przykład substancja P, wazopresyna czy też różne przeciwciała monoklonalne (mAb). Wzrasta zainteresowanie naukowców wcelowaną terapią antynowotworową z udziałem emiterów beta minus, gdyż jest to technika bardzo precyzyjna i dająca szerokie spektrum zastosowań.
Autor: Karolina Wójciuk
Literatura:
1. Gottesman, I.S., Mandarino, L.J., Gerich, J.E., 1982. Somatostatin: its role in health and disease. Spec. Top. Endocrinol. Metab. 4, 177-243
2. Bakker, W.H., Albert, R., Bruns, C., Breeman, W.A., Hofland, L.J., Marbach, P., Pless, J., Pralet, D., Stolz, B., Koper, J.W., Lamberts, S.W.J., Visser, T.J., Lamberts, E.P., 1991. [111In-DTPA-D-Phe1]-octreotide, a potential radiopharmaceutical for imaging of somatostatin receptor-positive tumors: synthesis, radiolabeling and in vitro validation. Life Sci. 49, 1583-1591
3. Reubi, J.C., Waser, B., van Hagen, M., Lamberts, S.W., Krenning, E.P., Gebbers, J.O., Laissue, J.A., 1992. In vitro and in vivo detection of somatostatin receptors in human malignant lymphomas. Int. J. Cancer 50, 895-900
4. Spradau, T.W., Edwards, W.B., Anderson, C.J., Welch, M.J., Katzenellenbogen, J.A., 1999. Synthesis and biological evaluation of Tc-99m-cyclopentadienyltricarbonyltechnetium-labeled octreotide. Nucl. Med. Biol. 26(1), 1-7.
5. Breeman, W.A., van der Wansem, K., Bernard, B.F., van Gameren, A., Erion, J.L., Visser, T.J., Krenning, E.P., de Jong, M., 2003. The addition of DTPA to [177Lu-DOTA0,Tyr3]octreotate prior to administration reduces rat skeleton uptake of radioactivity. Eur. J. Nucl. Med. Mol. Imaging 30, 312-315.
6. D'Huyvetter, M., Aerts, A., Xavier, C., Vaneycken, I., Devoogdt, N., Gijs, M., Impens, N., Baatout, S., Ponsard, B., Muyldermans, S., Caveliers, V., Lahoutte, T., 2012. Development of 177Lu-nanobodies for radioimmunotherapy of HER2-positive breast cancer: evaluation of different bifunctional chelators. Contrast. Media. Mol. Imaging 7(2), 54-64
7. Smith-Jones, P.M., Stolz, B., Albert, R., Ruser, G., Briner, U., Mäcke, H.R., Bruns, C., 1998. Synthesis and characterisation of [90Y]-Bz-DTPA-oct: a yttrium-90-labelled octreotide analogue for radiotherapy of somatostatin receptor-positive tumours.Nucl. Med. Biol. 25(3), 181-8
8. Arano, Y., Akizawa, H., Uezono, T., Akaji, K., Ono, M., Funakoshi, S., Koizumi, M., Yokoyama, A., Kiso, Y., Saji, H., 1997. Conventional and high-yield synthesis of DTPA-conjugated peptides: application of a monoreactive DTPA to DTPA-D-Phe1-octreotide synthesis. Bioconjug. Chem. 8(3), 442-6
Recenzje