



- Biochemia
- Biofizyka
- Biologia
- Biologia molekularna
- Biotechnologia
- Chemia
- Chemia analityczna
- Chemia nieorganiczna
- Chemia fizyczna
- Chemia organiczna
- Diagnostyka medyczna
- Ekologia
- Farmakologia
- Fizyka
- Inżynieria środowiskowa
- Medycyna
- Mikrobiologia
- Technologia chemiczna
- Zarządzanie projektami
- Badania kliniczne i przedkliniczne




Metody wykorzystywane do odbiałczania krwi, cz. I
 Osocze krwi jest ważnym środkiem transportującym metabolity w organizmie ssaków, a analiza chemiczna surowicy może dostarczyć wielu informacji odnoszących się do stanu biochemicznego osobnika i jest ona istotna dla celów diagnostycznych. Osocze jest bardzo złożone pod względem fizykochemicznym, ponieważ składa się z szeregu organicznych i nieorganicznych składników o szerokim zakresie mas cząsteczkowych i klas chemicznych, co sprawia, że analiza osocza nie jest łatwa. Coraz częściej do analizy osocza krwi poza tradycyjnymi metodami wykorzystuje się spektroskopię NMR (spektroskopia magnetycznego rezonansu jądrowego), która dostarcza bardzo użytecznych w diagnostyce informacji jakościowych i ilościowych związanych z zaburzeniami metabolicznymi [16].
Osocze krwi jest ważnym środkiem transportującym metabolity w organizmie ssaków, a analiza chemiczna surowicy może dostarczyć wielu informacji odnoszących się do stanu biochemicznego osobnika i jest ona istotna dla celów diagnostycznych. Osocze jest bardzo złożone pod względem fizykochemicznym, ponieważ składa się z szeregu organicznych i nieorganicznych składników o szerokim zakresie mas cząsteczkowych i klas chemicznych, co sprawia, że analiza osocza nie jest łatwa. Coraz częściej do analizy osocza krwi poza tradycyjnymi metodami wykorzystuje się spektroskopię NMR (spektroskopia magnetycznego rezonansu jądrowego), która dostarcza bardzo użytecznych w diagnostyce informacji jakościowych i ilościowych związanych z zaburzeniami metabolicznymi [16].
Osocze jest mieszaniną lipoprotein, białek, małych cząsteczek organicznych i janów, które razem uczestniczą w rozmaitych interakcjach, włączając tworzenie kompleksów metali, procesy wymiany chemicznej czy biotransformację enzymów [16].
W celu oznaczenia niebiałkowych składników krwi bądź surowicy, wykorzystywana jest odbiałczona krew (przesącz pozbawiony białka). W zależności od tego, jaki składnik krwi będzie oznaczany stosuje się różne metody odbiałczania, w których pod uwagę brane są takie parametry jak: pH przesączu, a także możliwość strącenia się z białkami oznaczanych związków.
Tak więc, dobór odpowiedniej metody odbiałaczania ma bardzo duży wpływ na dalsze wyniki przeprowadzanych oznaczeń. W trakcie doboru metody należy mieć również na uwadze, że przejście do przesączu np. związków redukujących może powodować podwyższenie wyników w metodach opartych na wykorzystaniu właściwości redukujących oznaczanej substancji [1].
Odbiałczanie jest niezbędnym krokiem stosowanym w wielu procedurach chemicznej analizy płynów ustrojowych. Do odbiałcznia krwi używa się wielu różnych odczynników, a najczęściej stosowany jest kwas wolframowy oraz kwas trójchlorooctowy [14]. W swoich doświadczeniach Golin i Wu (1919) wprowadzili kwas wolframowy do przygotowania filtrów niewiążących białek, które wykorzystywane były w stworzonej przez nich metodzie analizy krwi. W zaproponowanej metodzie używano 667 m.Eq (milirównoważnik) kwasu do wytrącenia białka z 1 litra osocza lub całej krwi oraz podwójnej objętości do odbiałczenia takiej samej ilości upakowanych krwinek czerwonych [14].
Dzięki zastosowaniu wolframianu sodu i kwasu siarkowego do przesączu dodawano równoważne ilości NA i SO4. Kwas trichlorooctowy powszechnie stosowany jest w 5% stężeniach w filtracie (przesączu)i nie może być stosowany w mniej niż połowa tego stężenia. Z tych względów kwas wolframowy i trichlorooctowy są nieprzydatnymi środkami odbiałczającymi w wielu metodach analitycznych i wielu innych nowoczesnych technikach. Tak więc, w trakcie opracowywania metod odbiałczania poszukiwano takich środków odbiałczających, które dodatkowo nie będą wprowadzać obcych substancji do filtratu i takich, które mogą być szybko zastosowane i powodować będą minimalne zniekształcenia obrazu chemicznego badanych płynów [14].
Golin i Wu (1919) podjęli próbę w poszukiwaniu właśnie takich substancji, a następnie opisali metodę otrzymywania wolnych od białek przesączów krwi, co również stosowano do oznaczania innych płynów ustrojowych. W zależności od badanego płynu w metodach strącania białek należy odpowiednio regulować pH rozcieńczonego płynu, co ma wpływ na szybkość strącania białek w trakcie ogrzewania roztworu. Osocze, komórki i pełna krew wymagają ok. 50, 27 i 40 milirównoważników (m.Eq) kwasu octowego/L odpowiednio do dostosowania pH. Uzyskane przesącze po krótkim ogrzewaniu w temperaturze 100°C są klarowne, bezbarwne i pozostają jasne z dodatkiem kwasu sulfosalicylowego [14].
Odbiałczanie krwi (wg Hunter G., 1956) [14].
Regulacja pH: w przedstawionej metodzie rutynowo stosowany był 0,05M kwas octowy, aczkolwiek mogą być wykorzystywane również inne kwasy. Jedna objętość osocza, komórek i pełnej krwi wymagała dodania odpowiednio: 1,0; 0,54 i 0,80 objetości kwasu octowego. Woda dodawana była do próbek do odpowiedniego rozcieńczenia np. 1 do 5 lub 1do 50- całość próbki dokładnie wymieszano.
Wytrącanie białek (wg Hunter G., 1956) [14].
W przypadku, gdy objętość roztworu jest mniejsza niż 10 ml wygodnie jest wykonywać doświadczenie w probówce wirówkowej. Badaną próbkę zanurzano na 3-5 minut w gotującej się wodzie, a następnie ochładzano. Ochłodzoną próbkę poddawano wirowaniu, a otrzymany po wirowaniu supernatant przenoszono do czystejj probówki. W przypadku większych objętości próbki badanej, czas ogrzewania musi być proporcjonalnie wydłużony (ogrzewanie w 100°C przez 2-4 minut) [14].
Sakuma R. i wsp.(1987) w swojej pracy opisali kilka metod odbiałczania próbek , które opierały się na wykorzystaniu m.in. wolframianu sodu, kwasu trichlorooctowego czy kwasu nadchlorowego [15].
Przygotowanie próbek do badań (wg Sakuma R. i wsp., 1987)
Próbki krwi żylnej pobrano do szklanych probówek nie zawierających antykoagulantu. Następnie, próbki pozostawiono w celu odstania na 60 minut w temperaturze pokojowej,a po upływie tego czasu odwirowano je (2000 xg przez 10 minut).
Następnie, przygotowano standard kwasu moczowego (300µmol/L) przez rozpuszczenie w wodzie destylowanej stocku kwasu moczowego (3 mmol/L w 8,12 mmol/L roztworze węglanu litu). Przygotowany w ten sposób standard przechowywano w temperaturze -20°C [15]. Bez względu na to jaką metodą przeprowadzano odbiałcznie (deproteinizację) próbek surowicy, standard zawsze przygotowywany był w ten sposób [15].
Metoda z wodorotlenkiem cynku (wg Sakuma R. i wsp., 1987)
Do mieszaniny 0,1 ml próbki surowicy oraz 0,5 ml (20g/L) siarczanu cynku dodano 0,5 ml 0,12mol/L roztworu hydratu baru zgodnie z procedurą opisaną przez Somogyi M. (1945). Tak przygotowaną próbkę mieszano przez vorteksowanie przez 10 s, a następnie odwirowano (2000 x g, 5 minut). Otrzymany supernatant rozpuszczono w dwóch objętościach buforu fosforanu sodu (40 mmol/L, pH=2.2) [15].
Metoda z wolframianem sodu (wg Sakuma R. i wsp., 1987)
0,1 ml próbki surowicy dodano do 1 ml roztworu wolframianu sodu (10g/L) oraz 67 mmol/L kwasu siarkowego (zgodnie z procedurą opisaną przez Haden RL. (1923). Próbke vorteksowano przez 10 s, a następnie odstawiono na 5 minut, po czym mieszaninę zwirowano (2000 x g, 5 minut). Otrzymany supernatant rozpuszczono dwukrotnie przy użyciu buforu fosforanu sodu [15].
W metodzie z trichlorooctanem (50g/L kwasu trichlorooctowego) oraz w metodzie z nadchloranem ( 0,4 mol/L roztworu nadchloranu), zastosowano identyczną procedurę jak z użyciem wolframianu sodu, z tym wyjątkiem, że otrzymany po wirowaniu supernatant rozpuszczono odpowiednio 0,2 mol/L lub 0,25 mol/L wodorofosforanu sodu [15].
Odbiałczanie metodą z acetonitrylem: 0,2 ml surowicy zmieszano z róną objętością acetonitrylu, a następnie próbkę zwirowano przy 2000 x g przez 5 minut. Następnie, 0,1 ml supernatantu otrzymanego po wirowaniu rozpuszczono z 0,1 ml fosforanu sodu [15].
Do określenia stężenia białka otrzymanego powyższymi metodami zastosowano metodę Lowrye’go. Przeprowadzone doświadczenia wykazały, że wykorzystane metody pozwalają na odbiałczanie próbek z dużą wydajnością- usuwanie od 98% do 100% białek z badanych próbek. Metody odbiałczania z acetonitrylem oraz nadchloranem są mniej wydajne w porównaniu do pozostałych, opisanych w pracy (wg Sakuma R. i wsp., 1987) [15].
Wybrane metody odbiałczania krwi
1) Odbiałczanie z wykorzystaniem kwasu trichlorooctowego
Kwas trichlorooctowy strąca tylko białka, w przypadku, gdy jego końcowe stężenie nie jest mniejsze od 3%. Otrzymany po strącaniu przesącz posiada właściwości silnie kwasowe.
Wykonanie:
Do 2 ml surowicy (lub krwi) należy dodać 8 ml 10% roztworu kwasu trichlorooctowego (CCl3COOH). Całą próbkę bardzo dokładnie wymieszać , a następnie odwirować przez 10 minut bądź poddać przesączeniu.
2) Odbiałczanie z wykorzystaniem koagulacji na gorąco
Do 2 ml surowicy dodać 2 ml wody oraz 2 ml 3% roztworu kwasu octowego. Tak przygotowaną próbkę inkubować przez 5 minut we wrzącej łaźni wodnej, zaś po upływie czasu inkubacji należy ją odwirować.
W latach 1920 oraz 1925, wprowadzono pierwszą klasyfikację lipidów (Bloor), kiedy to opisano lipidy jako dużą klasę związków biologicznych obejmujących kwasy tłuszczowe oraz ich naturalne pochodne. Wykorzystywany do odbiałczania odczynnik Bloora stanowi mieszaninę etanolu i eteru w stosunku 3:1. Odczynnik ten wykorzystywany jest w oznaczaniu w surowicy krwi tłuszczów, które to ekstrahowane są za pomocą eteru i etanolu [1], [2].
3) Odbiałczanie mieszaniną Bloora
Wykonanie:
Do kalibrowanej probówki o objętości 10 ml należy dodać 9 ml mieszaniny Bloora oraz 1 ml surowicy. Zawartość probówki dokładnie wytrząsnąc, a dalej wstawić ją do łaźni wodnej o temperaturze 60°C- 70°C. Probówkę poddać kilkunasto minutowej inkubacji. Po inkubacji, próbkę uzupełnić do objętości 10 ml za pomocą mieszaniny Bloora, całość dokładnie wymieszać, po czym próbkę odwirować lub przesączyć [1].
Ekstrakcja lipidów z erytrocytów (Rodriguez-Vico F. i wsp., 1991)
Poniższa metoda ekstrakcji lipidów z erytrocytów umożliwia równoczesne oznaczanie białek w pozostałości poekstrakcyjnej. Jako materiał badany nalezy uzyć zawiesiny krwinek czerwonych , przemytych za pomoca buforu PBS [3].
Wykonanie:
a)250 µl upakowanych krwinek należy umieścić w dużej probówce lub zlewce, ciągle mieszać przy wytrząsaniu z 25 ml mieszaniny heksanu z izopropanolem (zmieszanych w stosunku 3:2).
b)Próbkę odwirować przez 15 minut przy 4000 x g
c)Otrzymany po wirowaniu sypernatant przenieść ilościowo za pomoca pipetki pasterowskiej do suchej kolbki do wyparki prózniowej
d)Następnie, należy odparować rozpuszczalnik w wyparce przy maksymalnej temperaturze do 40°C
e)Otrzymaną suchą pozostałość lipidową rozpuścić w 200 ml mieszaniny chloroform:metanol (w stosunku 2:1), otrzymaną w ten sposób próbkę przechowywać w temperaturze -70°C [2],[3].
Do związków azotowych krwi, oprócz białek, należą także niebiałkowe związki azotowe, zwane resztą azotową lub azotem pozabiałkowym. Wszystkie te związki transportowane są przez krew, gdzie następnie jako tzw. związki niskodrobinowe przechodzą do moczu. W ten sposób są one następnie wydalane z ustroju. Do głównych niebiałkowych związków azotowych (usuwanych z ustroju) należą mocznik i kwas moczowy.
Mocznik jest końcowym produktem przemiany azotu białkowego w ustroju człowieka. Związek ten wytwarzany jest w wątrobie w procesie tzw. ureogenezy- powstaje z NH3 i CO2. W warunkach fizjologicznych stężenie mocznika we krwi plasuje się w granicy od 21 do 53 mg/dl. Cały proces biosyntezy mocznika w organizmie nazywany jest cyklem mocznikowym [4].
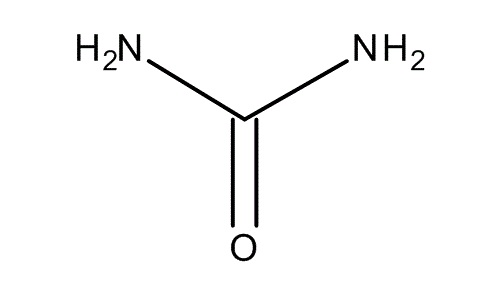
Zdjęcie: mocznik-wzór, http://www.merckmillipore.com/INTL/en/product/poland/chemicals/mocznik,MDA_CHEM-108486, [8].
Kreatyna (kwas N-metyloguanidynooctowy)jest związkiem powstającym w wątrobie, w wyniku transamidynacji glicyny. Cały proces zachodzi przy udziale argininy. Kreatynina transportowana jest do mięśni, gdzie łączy się z resztą fosforanową ATP- przechodzi w fosfokreatynę (określaną również mianem fosfagenu). Prawidłowa wartość kreatyniny w osoczu wynosi od 0,7 do 1,5 mg/dl. Kreatyna przekształcona w bezwodnik kreatyny (tzw. kreatyninę) wydalana jest z organizmu wraz z moczem [4]. Wolna kreatynina i fosfokreatynina w mięśniach jest nieenzymatycznie przekształcana do kretyniny, która dyfunduje do krwi i następnie jest wydalana przez nerki [17]. Ponieważ nerki odpowiedzialne są za usuwanie kreatyniny z krwi, pomiar stężenia kreatyniny w surowicy jest przydatnym wskaźnikiem funkcji nerek. Choć poziom kreatyniny różni się w zależności od stopnia uszkodzenia, badania wykazały, że u pacjentów cierpiących na niewydolność nerek zwykle zaczyna się on od 2mg/dl. Ponadto, nieprawidłowe kreatyniny moga wskazywać na rozwój cukrzycy czy chorób układu krążenia [17].
Odczyn Weyla
Do 2 ml roztworu kreatyniny dodać 4-5 kropli nasyconego roztworu nitroprusydku sodu oraz 1 ml 10% roztworu wodorotlenku sodu (NaOH). W probówce dochodzi do zmiany koloru roztworu na czerwony. Po dodaniu roztworu kwasu octowego czerwony kolor znika. Oznaczanie kreatyniny w surowicy krwi może powodować barwną reakcję przez reakcję występujących w niej substancji ketonowych, jednakże czerwone zabarwienie roztworu utrzymuje się jeszcze po dodaniu kwasu octowego [18].
Ilościowe oznaczanie kreatyniny metodą Folina-Wu
Kreatynina powstaje w wyniku przemiany materii (głównie mięśni szkieletowych). Jej stężenie oznacza się zarówno we krwi, jak i w moczu. Ze względu na wydalanie przez nerki, oznaczanie kreatyniny służy do oceny wielkości przesączania kłębuszkowego (tzw. GFR), które jest jednym z najważniejszych wskaźników czynności nerek [6].
Zasada metody oznaczania krestyniny z wykorzystaniem procedury Folina-Wu opiera się na fakcie, iż kreatynina wraz z kwasem pikrynowym tworzy kompleks o zabarwieniu pomarańczowo-czerwonym. Powstający związek oznaczany jest następnie z wykorzystaniem metody kolorymetrycznej [4].
Autor: Lidia Koperwas
Literatura:
[1]. Kłyszejko-Stefanowicz L, 2003. Ćwiczenia z biochemii. Wydawnictwo Naukowe PWN, 2003, s.580-582, 592-595.
[2]. http://www.ibmb.uni.wroc.pl/studia/2.pdf
[3]. Rodriguez-Vico F., Martinez-Cayuela M., Zafra M.F., Garcia-Peregrin E., Ramirez H., 1991. Lipids 26 (77-80).
[4]. Niebiałkowe związki azotowe -mocznik, kwas moczowy, kreatynina; ilościowe oznaczanie kreatyniny metodą Folina-Wu. Ćwiczenie nr 13. http://farmacja.cm-uj.krakow.pl/public_includes/upload/bioch_farm_cwiczenie_nr_13.pdf
[5]. Posadzy-Małaczyńska A., Tykarski A., 2011. Kwas moczowy w chorobach sercowo-naczyniowych – co nowego?. Borgis - Postępy Nauk Medycznych s3/2011, s. 46-50. http://www.pnmedycznych.pl/shown.php?ktory=4061
[6]. Drabczyk R., 2014. Kreatynina. Medycyna praktyczna dla pacjentów. http://nefrologia.mp.pl/diagnostyka/show.html?id=51973
[7]. http://www.merckmillipore.com/PL/en/product/Creatinine,MDA_CHEM-105206
[8]. http://www.merckmillipore.com/INTL/en/product/poland/chemicals/mocznik,MDA_CHEM-108486
[9]. Majdan M., Borys O., 2010. DNA i schorzenia towarzyszące podwyższonemu stężeniu kwasu moczowego. ANNALES ACADEMIAE MEDICAE STETINENSIS ROCZNIKI POMORSKIEJ AKADEMII MEDYCZNEJ W SZCZECINIE 2010, 56, SUPPL. 1, 34–39. http://www.pum.edu.pl/__data/assets/file/0009/29817/SUPLEMENT_56-01_05.pdf
[10]. Januszkiewicz-Caulier J., Franek E., 2011. Kwas moczowy w chorobach nerek, serca i naczyń. 31
K L I N I C Z N A I N T E R P R E T A C J A W Y N I K Ó W B A D A Ń. Choroby Serca i Naczyń 2011, tom 8, nr 1, 31–37. http://czasopisma.viamedica.pl/chsin/article/viewFile/18633/14657
[11]. Oznaczanie mocznika w płynach ustrojowych metodą hydrolizy enzymatycznej, http://pchba.amu.edu.pl/cw%20CBA/cw3.pdf
[12]. Ayelet Erez MD, PhD, 2013. Argininosuccinic aciduria: from a monogenic to a complex disorder. Genetics in Medicine (2013) 15, 251–257, http://www.nature.com/gim/journal/v15/n4/fig_tab/gim2012166f3.html.
[13]. http://chemklin.sum.edu.pl/uploaded/Chemia%20kliniczna/Kwas%20moczowy.pdf
[14]. Hunter G., 1957. A method for deproteinization of blood and other body fluids. J.clin. Path. (1957), 10, 161.
[15]. Sakuma R., Nishina T., Kitamura M., 1987. Deproteinizing Methods Evaluated for Determination of Uric Acid in Serum by Reversed –Phase Liquid Chromatography with Ultraviolet Detection. Clin. Chem. 33 No 8, 1427-1430 (1987). [16]. Daykin CA, Foxall PJ, Connor SC, Lindon JC, Nicholson JK. The comparison of plasma deproteinization methods for the detection of low-molecular-weight metabolites by (1)H nuclear magnetic resonance spectroscopy. Anal Biochem. 2002 May 15;304(2):220-30. http://www.ncbi.nlm.nih.gov/pubmed/12009699
[17]. https://www.caymanchem.com/pdfs/700460.pdf
[18]. http://usfiles.us.szc.pl/pliki/plik_1262018104.pdf
[19]. http://www.wiener-lab.com/wiener/catalogo/archivos/6322_creatinina_enzimatica_aa_liquida_pl.pdf
[20]. http://www.diag.pl/Badanie-Klirens-kreatyniny.85+M51588beb2ff.0.html
[21]. Fossati P., Prencipe L., 2010. Chromogenic System for Measuring Hydrogen Peroxide: The Enzymatic Uric Acid Assay. Clinical Chemistry May 2010 vol. 56 no. 5 865-866. http://www.clinchem.org/content/56/5/865.full
[22]. http://chemklin.sum.edu.pl/uploaded/kreatynina.pdf
[23]. Bugajska J., Berska J., Hodorowicz-Zaniewska D., Sztefko K., 2010. Walidacja metody oznaczania kwasów tłuszczowych frakcji fosfolipidów w surowicy krwi. diagnostyka laboratoryjna, Journal of Laboratory Diagnostics 2010 • Volume 46 • Number 2 • 125-130. http://diagnostykalaboratoryjna.eu/journal/DL_2_2010._str_125-130.pdf
[24]. Bezpośrednia metoda kolorymetryczna do oznaczania żelaza w surowicy krwi lub osoczu. Fer-color, Wiener-lab. http://www.wiener-lab.com/wiener/catalogo/archivos/12055_fer_color_aa_liquida_pl.pdf
[25]. Elmagirbi A., Sulistyarti H., Atikah, . Study of Ascorbic Acid as Iron (III) Reducing Agent for Spectrophotometric Iron Speciation. J. Pure App. Chem. Res., 2012, 1(1), 11-17. http://jpacr.ub.ac.id/index.php/jpacr/article/view/101/98
Recenzje