



- Biochemia
- Biofizyka
- Biologia
- Biologia molekularna
- Biotechnologia
- Chemia
- Chemia analityczna
- Chemia nieorganiczna
- Chemia fizyczna
- Chemia organiczna
- Diagnostyka medyczna
- Ekologia
- Farmakologia
- Fizyka
- Inżynieria środowiskowa
- Medycyna
- Mikrobiologia
- Technologia chemiczna
- Zarządzanie projektami
- Badania kliniczne i przedkliniczne




Wybrane metody oznaczania azotanów (V) i (III) w wodzie i produktach spożywczych cz.II
Największym źródłem azotanów w żywności są warzywa. Ich obecność jest zjawiskiem naturalnym wynikającym z procesu nawożenia azotowego roślin. Z racji tego, że azotany są szybko wydalane z organizmu, stanowią niewielkie zagrożenie dla człowieka. Ich szkodliwość wzrasta dopiero po redukcji do azotanów (III), co zazwyczaj ma miejsce po zbiorze warzyw, a także w przewodzie pokarmowym człowieka. Zawartość azotanów (III) i azotanów (V) w produktach spożywczych zależy od kilku czynników: gatunku rośliny,czynników środowiskowych,nawożenia azotowego, typu gleby czy terminu zbioru [1], [4].
Azotany (V) gromadzą się głównie w warzywach korzeniowych np. burak ćwikłowy, marchwi oraz w warzywach liściastych o krótkim okresie wegetacji, w tym np. w szpinaku [4]. Z kolei, azotany (III) w warzywach mogą tworzyć się podczas składowania i przechowywania w zamkniętych opakowaniach foliowych. Najczęściej powstają w warzywach przechowywanych w pomieszczeniach, które są niechłodzone lub w warzywach uszkodzonych (na skutek uszkodzenia tkanki) z zachodzącymi procesami gnilnymi [4]. Największe ilości azotanów występują w młodych roślinach. Jednym ze sposób zmniejszania zagrożeń zdrowotnych jest rozwój rolnictwa ekologicznego i zmiana nawyków żywieniowych. Co ciekawe, pomimo mniejszych ilości zanieczyszczeń chemicznych w roślinach pochodzących z gospodarstw ekologicznych niż konwencjonalnych, w porównaniu z dietą tradycyjną większe ilości azotanów wykrywane są w diecie wegetariańskiej [1].
Azotany (V) i (III) przedostają się do organizmu człowieka głównie z żywnością i wodą do picia. Związki te wpływają na wykorzystanie witamin, białek, węglowodanów i tłuszczów zawartych w pożywieniu [3]. W wodach powierzchniowych azotany występują zwykle w niewielkich stężeniach, stanowiąc najwyższy stopień utlenienia związków organicznych i nieorganicznych. W znacznych ilościach azotany identyfikowane są także w ściekach po biologicznym oczyszczeniu, z kolei obecność azotanów wodach powierzchniowych wiąże się z doprowadzeniem ze ścieków miejskich i przemysłowych, z odwodnień kopalń bądź wskutek spływu wody z pól, na których stosowano sztuczne nawożenie [8].
Droga azotanów i azotynów w organizmie
Przeprowadzone badania potwierdziły, że w oparciu o jonowe mechanizmy transportu aniony azotanowe oraz azotynowe mogą być wchłaniane w żołądku, jelicie, tarczycy oraz gruczołach ślinowych. Po wchłonięciu do krwi azotany oraz azotyny transportowane są do wątroby, gdzie ulegają utlenianiu do azotanów i w tej postaci zostają wydalone z organizmu. W przypadku np. infekcji bakteryjnej pęcherza moczowego, azotany i azotyny wydalane są z moczem w postaci azotynów. Jest jedyny przypadek kiedy to jon azotynowy może być wydalony z organizmu. Azotany i azotyny wchłonięte do organizmu ssaków mogą ulegać recylkulacji w następstwie przeniknięcia z krwi do gruczołów ślinowych, następnie ze śliną związki te ponownie przedostają się do odcinka żołądkowo-jelitowego w przewodzie pokarmowym, gdzie ulegają ponownemu wchłanianiu. Azotyny obok związków N-nitrozowych zostały sklasyfikowane jako czynniki, które zwiększają ryzyko raka żołądka i raka jelita grubego u ludzi [2].
Tlenek azotu (NO)
Analiza jonów NO2- i NO3- powstających w metabolizmie tlenku azotu, pozwala na badania nad jego uczestnictwem w wielu fizjologicznych i patofizjologicznych procesach, takich jak rozkurczanie mięśni gładkich,regulacje immunologiczne organizmu, arterioskleroza, nadciśnienie tętnicze czy cukrzyca. Tlenek azotu powstaje w organizmach żywych w małych ilościach i bardzo szybko zostaje utleniony do azotynów i azotanów. Dlatego też, bezpośrednie oznaczenie NO jest problematyczne, a jednym z elementów badań metabolizmu tlenku azotu w próbkach tkanek jest określanie stosunku NO2- /NO3- [5].
Tlenek azotu (NO) produkowany jest w organizmach zwierząt przez 3 odrębne formy enzymu znanego jako syntaza tlenku azotu (NOS). Tak więc, w organizmie powstają dwie konstytutywne formy tlenku azotu: neuronalna (nNOS — NOS I) i śródbłonkowa (eNOS — NOS III), które zależne są od jonów wapnia i stymulowane są za pośrednictwem kalmoduliny oraz tzw. forma indukowana (iNOS — NOS II) [6].
Tlenek azotu a kancerogeneza
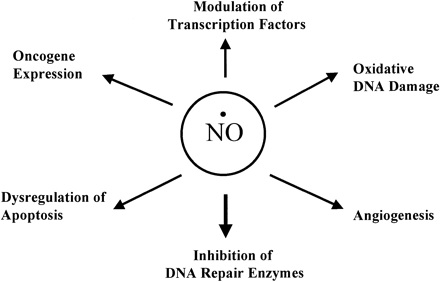
Zdjęcie: Rola tlenku azotu (NO) w karcinogenezie, http://ajpgi.physiology.org/content/281/3/G626
Obecność azotanów w wodach pitnych musi być ograniczona, ponieważ mogą one powodować wiele niekorzystnych zjawisk, w tym sinicę u niemowląt (methemoglobinemia) czy nowotwory układu pokarmowego u ludzi i zwierząt. W związku z tym, zawartość azotynów w wodach do picia musi być ściśle kontrolowana. Jednocześnie azotany i azotyny są wykorzystywane jako dodatki do produktów żywnościowych, które odpowiadają m.in. za zabezpieczenie żywności przed skażeniem biologicznym. Niestety związki te mogą też reagować in vivo z aminami tworząc kancerogenne nitrozaminy, co z kolei wymusza określanie zawartości azotynów i azotanów w żywności, podobnie jak w wodzie do picia. Powszechne występowanie w środowisku jonów NO2- i NO3- sprawia, że monitorowanie ich stężeń w próbkach wody jest dość trudne [5].
Analiza azotanów w wodzie
Wśród głównych metod oznaczania jonów NO2- i NO3- znajdują się te oparte na technikach spektroskopowych (podobnie jak klasyczna metoda Griessa). Niestety, procedury te są czasochłonne, a dodatkowo bardzo podatne na różnego rodzaju interferencje. Ponadto mogą być niemożliwe do zastosowania dla niektórych próbek żywności i próbek biologicznych, gdyż często wiąże się to z problemami związanymi z otrzymaniem klarownych roztworów do końcowego pomiaru. Problem stanowi też obecność innych jonów (np. chlorkowych) występujących zwłaszcza w próbkach biologicznych. Czułość metody w stosunku do jonów azotynowych jest stosunkowo niska, a śladowy poziom tych jonów w próbkach bardzo często jest niemożliwy do wykrycia. Dlatego też, bardzo przydatną metodą jest wysokosprawna chromatografia cieczowa (HPLC). HPLC jest metodą szybszą, czulszą i bardziej selektywną niż metody oparte na oznaczeniach kolorymetrycznych [5]. Na przestrzeni kilku lat opracowano wiele metod rozdzielania i detekcji anionów nieorganicznych z wykorzystaniem chromatografii jonowymiennej lub chromatografii odwróconych faz i par jonowych przy wykorzystaniu detekcji anionów za pomocą detektora konduktometrycznego, odwróconej detekcji UV, absorpcyjnych detektorów promieniowania UV czy detektorów chemiluminescencyjnych i detektorów elektrochemicznych. Analizę azotanów w wodzie pitnej najczęściej przeprowadza się stosując wysokosprawną chromatografię jonowymienną (IC-HCLP) [5].
Oznaczanie zawartości azotanów (V) i (III) metodą kolorymetryczną w próbkach pochodzenia roślinnego
W metodzie tej wykorzystywany jest odczynnik Griessa, a pomiar wykonywany jest przy długości fali równej λ=538 nm. W reakcji wykorzystywana jest bezpośrednia redukcja azotanów (V) do azotanów (III) za pomocą metalicznego kadmu. Na początkowym etapie oznaczania, próbę wytrząsa się z węglem aktywnym, w celu usunięcia zabarwienia pochodzącego z chlorofilu. W trakcie doświadczenia dokonuje się pomiaru pH w zhomogenizowanej miazdze (próbka roślinna) z wykorzystaniem pH-metru. Oznaczanie ekstraktu ogólnego wykonuje się metodą refraktometryczną [4].
Metoda kolorymetrycznego oznaczania azotanów w próbkach wody
Zasada metody:
W środowisku stężonego kwasu siarkowego jony azotanowe ulegają reakcji z salicylanem sodu. Produktem reakcji jest mieszanina kwasów: 3-nitrosalicylowego i 5-nitrosalicylowego, których sole w środowisku alkalicznym wykazują żółte zabarwienie. Bez konieczności rozcieńczania (bądź zatężania) próbki wody za pomocą metody kolorymetrycznej można oznaczać azotany znajdujące się w stężeniach: od 0,02 –4,5 mg/dm3 NNO3. W prawidłowym oznaczaniu azotanów przeszkadzają: mętność znajdująca się powyżej poziomu 30 mg/dm3 O2, żelazo w ilościach powyżej 5 mg/dm3 Fe oraz chlorki w stężeniach powyżej 200 mg/dm3 [9].
Przygotowanie roztworów wykorzystywanych podczas oznaczenia próbek wody
- Azotan (V) potasu (podstawowy roztwór wzorcowy)
0,7216 g azotanu potasu wysuszonego wcześniej w 105°C do stałej masy należy rozpuścić w wodzie destylowanej,a dalej dodać 1 cm3 chloroformu. Kolbę dopełnić wodą destylowaną do 1 dm3. W 1 cm3 tak przygotowanego roztworu zawarte jest 0,1 mg azotu azotanowego (V) [9].
- Azotan (V) potasu (roboczy roztwór wzorcowy)
Z podstawowego roztworu wzorcowego azotanu (V) potasu pobrać 10 cm3 do parownicy, dodać 2-3 krople 0,5 % roztworu NaOH i 20 cm3 roztworu salicylanu sodu. Odparować mieszaninę w parownicy do sucha na łaźni wodnej. Do suchej pozostałości dodać 1 cm3 roztworu H2SO4 (rozprowadzić go po ściankach w miejscach z białym osadem). Inkubować 10 minut, a dalej do parownicy dodać ok. 30 cm3 wody destylowanej. Całość dokładnie wymieszać i przenieść ilościowo do kolby miarowej o poj. 100 cm3 (dopełnić wodą destylowaną do kreski). Przygotowany roztwór w 1 cm3 zawiera 0,01 mg azotu azotanowego (V) [9].
- Przygotowanie skali wzorców
Przygotować 10 kolb miarowych, do których kolejno odmierzyć odpowiednie ilości roboczego roztworu wzorcowego azotanu (V) potasu, tj.: 0,0; 0,2; 0,3; 0,5; 0,7; 1,0; 2,0; 3,0; 4,0 i 5,0 cm3. Do każdej z kolb odmierzyć po 7 cm3 alkalicznego roztworu winianu i potasu. Całość dopełnić wodą destylowaną do kreski i dokładnie wymieszać . Przygotowane wzorce zawierają następujące ilości NNO3: 0,00; 0,002; 0,003; 0,005; 0,007; 0,01; 0,02; 0,03; 0,04; 0,05 mg.
Każdy z roztworów przelać kolejno do kuwety i dokonać pomiaru absorbancji (przy długości fali λ= 436 nm). W trakcie wykonywania oznaczenia jako odnośnik należy wykorzystać roztwór wzorcowy (przygotowany jako pierwszy – pierwsza kolba) [9].
W trakcie pomiaru absorbancji w zależności od wyników zawartości azotanów w próbce należy wykorzystywać kuwety pomiarowe o różnej grubości warstwy absorpcyjnej, i tak:
- przy zawartości NNO3 znajdujących się w granicach od 0,002 –0,007 mg w próbce należy stosować kuwety o grubości warstwy absorpcyjnej równej 5 cm,
- przy zawartości w granicach 0,01 –0,5 mg używa się kuwet o grubości warstwy absorpcyjnej równej 1 cm [9].
Wykonanie oznaczenia:
W parownicy porcelanowej umieścić taką ilość przygotowanej próbki wody, by zawartość w niej NNO3 mieściła się w granicach 0,002 –0,05 mg. Do próbki dodać 2 –3 krople 0,5% roztworu wodorotlenku sodu i 1 cm3 0,5% roztworu salicylanu sodu. Zawartość parownicy odparować do sucha na łaźni wodnej, pozostawić do ostygnięcia. Zwilżyć suchą pozostałość za pomocą 1 cm3 kwasu siarkowego (VI) (kwas należy rozprowadzić po ściankach parownicy w miejscach pokrytych osadem). Próbkę inkubować przez 10 minut, a dalej rozcieńczyć 20 cm3 wody destylowanej. Do próbki dodać 7 cm3 alkalicznego roztworu winianu sodu i potasu (tj.: 400 g NaOH i 60 g winianu sodu i potasu rozpuścić w wodzie destylowanej, ostudzić i rozcieńczyć do objętości 1 dm3 wodą destylowaną, dokładnie wymiesza). Całość przenieść ilościowo do kolby miarowej, uzupełnić wodą destylowaną do kreski i dokładnie wymieszać. Oznaczać absorbancję przy długości fali λ = 436 nm, stosując odpowiednią kuwetę, a jako odnośnik zastosować próbkę kontrolną (przygotowaną jak pierwszy wzorzec).
Zawartość azotu azotanowego (V) w badanej próbce należy odczytać z przygotowanej wcześniej krzywej kalibracyjnej, stanowiącej zależność zmierzonej absorpcji [AU] od ilości azotu azotanowego w próbce [mg].
Stężenie azotu azotanowego w wodzie obliczyć stosując poniższy wzór:
X= a * 1000 / V mg/dm3 NNO3
gdzie:
a - ilość azotu azotanowego w próbce określona na podstawie krzywej kalibracyjnej [mg]
V – objętość próbki wody użytej do oznaczania [cm3] [9].
Gotowy test do kolorymetrycznego oznaczania azotanów (wg instrukcji ze strony: http://www.perfectwater.com.pl/instrukcje/Testoval%20AZOTANY%20instrukcja.pdf)
Na rynku dostępne są gotowe testy do kolorymetrycznego oznaczania azotanów. Oznaczenie można wykonywać w zakresie od 0 do 20 mg/l NO3-. Testy te są niezwykle przydatne, ponieważ skracają całkowity czas pomiaru do kilku minut [10].
Wykonanie oznaczenia z wykorzystaniem gotowego zestawu odczynników (http://www.perfectwater.com.pl/instrukcje/Testoval%20AZOTANY%20instrukcja.pdf)
Szklaną probówkę napełnić badaną wodą do górnej podziałki o wartości 10 ml. Następnie dodać dwie czubate miarki (łyżeczka czerwona) odczynnika A oraz dwie pełne łyżeczki odczynnika B. Probówkę natychmiast zatkać korkiem i obracać probówkę przez 60 sekund (obracać ok. 30 razy probówką –korkiem w dół i do góry). Nie wstrząsać! Po upływie 1 minuty zamkniętą probówkę pozostawić na 6-minutową inkubację. Po tym czasie z kasetki kolorymetrycznej (dołączonej do zestawu) wyjąć kwadratowy pojemniczek, napełnić go wodą uprzednio wymieszaną z odczynnikami A i B, po czym pojemnik włożyć z powrotem do kasetki kolorymetrycznej (w celu porównania barw i odczytania wyniku)- w tym celu kasetkę ustawić pod światło lub na białym tle. Barwę ze środkowego pola należy porównać z polami zewnętrznymi, które oznaczone są różnymi wartościami. Wynikiem przeprowadzonego oznaczenia będzie wartość z pola zewnętrznego i najbardziej zbliżonego barwą do barwy pola środkowego [10].
W przypadku, gdy podczas przygotowywania próbki wody z odczynnikami w probówce pojawi się barwa mocno czerwona lub zmętnienie próbki oznacza to, że zawartość azotanów w badanej próbce przekracza zakres pomiarowy przyrządu [10]. W celu oznaczenia próbki należy postąpić następująco:
probówkę należy przemyć wodą destylowaną, po czym napełnić ją do wysokości 1 ml (na podziałce probówki) badaną próbką wody. Całą próbkę uzupełnić do poziomu 10 ml wodą destylowaną, po czym dodać odczynniki A i B (w ilości jak wyżej). Wszystkie pozostałe czynności wykonywać w identyczny sposób jak opisano powyżej [10]. W przypadku występowania w próbce ilości azotanów przekraczających zakres pomiarowy przyrządu wynik pomiaru należy pomnożyć przez liczbę 10 (wówczas oznaczy się właściwą zawartość azotanów w badanej próbce wody) [10].
Zespół wstrzykowych analizatorów przepływowych do równoległego oznaczania azotynów i azotanów (zestaw firmy MLE)
Oprócz gotowych zestawów odczynników na rynku dostępne są specjalnie zaprojektowane analizatory wykorzystywane do równoległego oznaczania azotynów i azotanów w próbce. Analizatory mają konstrukcję modułową i kompaktową, wyposażone są w ośmioportowe zawory wstrzykowe z dwiema pętlami o różnych pojemnościach oraz sześciokanałowe pompy perystaltyczne i fotometry z dwoma sensorami. Cała zaawansowana konstrukcja analizatora zapewnia dużą stabilność sygnału analitycznego. Wstrzykowe analizatory przepływowe pozwalają na oznaczenie w prosty, a zarazem równoległy sposób stężenia azotanów i azotynów mieszczące się w zakresie od 0,2 mg/L N-NO2 i N-NO3.
Zasada metody oznaczania azotynów polega na tworzeniu soli diazowej, która powstaje w wyniku reakcji azotynów z sulfanilamidem. Sól diazowa ulega sprzężeniu z N-(1-naftylo-)-etylenodiaminą w wyniku czego powstaje barwnik azowy. Oznaczanie azotanów w próbce polega na ich wcześniejszym oddzieleniu od matrycy na membranie dializatora, a następnie redukcji w kolumnie kadmowej do azotynów. Reakcja zachodzi w środowisku buforu imidazolowego w pH=7,5. Azotyny oznaczane są w sposób opisany powyżej [12].
Próba fenoloftaleinowa na wykrywanie HNO3 i jego soli [11].
Jony azotanowe NO3- odbarwiają fenoloftaleinę zabarwioną w kwasie siarkowym, z kolei sole kwasu azotowego przeprowadzają powyższą reakcję w obecności nadtlenku wodoru (H2O2). Wykorzystując tę zasadę możliwe jest oznaczanie śladowych zanieczyszczeń w kwasie siarkowym lub innych substancjach pochodzących od kwasu azotowego i azotynów, a oznaczenie można przeprowadzać w stosunku 1: 1500 000. Z wykorzystaniem tej próby możliwe jest również oznaczanie mniejszych zanieczyszczeń, co ujawnia się nie całkowitym, a częściowym odbarwieniem fenoloftaleiny [11].
Wykonanie:
1 g azotynu rozpuścić w 500 ml wody, a y otrzymanego roztworu pobrać 1 kroplę i wprowadzić do 100 ml stężonego kwasu H2SO4. Z tak zanieczyszczonego roztworu kwasu pobrać kolejne 4 ml (przed każdym pobraniem wytrząsnąć roztwór macierzysty) i dodać do nich 1 kroplę 0,05% alkoholowego roztworu fenoloftaleiny. Powstająca w próbce różowa barwa fenoloftaleiny znika po dodaniu 1 kropli 0,25% roztworu nadtlenku wodoru (H2O2) [11].
Stężony roztwór nadtlenku wodoru może i bez pomocy azotynu odbarwić fenoloftaleinę (np. kropla 15% H2O2 odbarwia ją po 2 godzinach) dlatego ważne jest dobranie takich rozcieńczeń nadtlenku wodoru, które nie mogą samodzielnie przeprowadzić opisanej reakcji. Czułość opisanej próby fenoloftaleinowej na wykrywanie kwasu HNO3 i jego soli wzrasta dzięki zastosowaniu w doświadczeniu H2O2. W tym celu najlepiej przygotowywać odczynnik do wykrywania śladowych zanieczyszczeń HNO3 i jego soli w H2SO4 wg przepisu: 50 ml 0,05% alkoholowego roztworu fenoloftaleiny zmieszać z 1 kroplą perhydrolu. Dzięki temu przeprowadzona próba jest czulsza od niespecyficznej próby dwufenyloaminowej, ponieważ 1 kropla odczynnika (odbarwienie) może wykryć w obj. 4 ml kwasu siarkowego około 0,00005% HNO3 lub jego soli. Niestety powyższe oznaczenie nie może być wykorzystywane do oznaczania azotynów i azotanów w substancjach zabarwiających stężony kwas siarkowy [11].
Oznaczanie azotynów i azotanów (wg Garbuliński T., 1955)
- Oznaczanie azotynów: do 4-5 ml chemicznie czystego roztworu kwasu siarkowego dodać 1 kroplę 0,05% alkoholowego roztworu fenoloftaleiny. W wyniku reakcji w próbce pojawi się różowe zabarwienie. Następnie do zabarwionej próbki kwasu siarkowego dodać kilka kropli substancji badanej na zawartość azotynów. Barwa w próbce znika po dodaniu 1 kropli 0,25% H2O2.
Jeżeli barwa w próbce zniknie przed dodaniem nadtlenku wodoru oznacza to, że w badanej substancji obecne są azotany [11].
- Oznaczanie azotanów: do 4-5 ml chemicznie czystego kwasu siarkowego znajdującego się w probówce dodać 1 kroplę 0,05% alkoholowego roztworu fenoloftaleiny z dodatkiem perhydrolu (tj.: 1 kropla perhydrolu w 50 ml fenoloftaleiny). Kwas zmienia zabarwienie na różowe, które znika po dodaniu do próbki kilku kropli próbki zawierającej w swoim składzie azotany (dodatek nadtlenku wodoru rozszerza zakres czułości próby na azotany) [11].
Jeżeli chcemy oznaczyć azotyny lub azotany w zanieczyszczonym kwasie siarkowym możliwe jest przeprowadzenie próby bezpośrednio na próbce zanieczyszczonego kwasu (nie jest konieczne stosowanie chemicznie czystego kwasu). Wówczas do 5 ml kwasu należy dodać 1 kroplę 0,05% alkoholowego roztworu fenoloftaleiny oraz 1 kroplę 0,25% roztworu H2O2. Większe stężenia azotanów w kwasie siarkowym blokują powstawanie różowego zabarwienia fenoloftaleiny. W przypadku obecności w próbce azotanów i azotynów pojawiające się w próbce zabarwienie można usunąć dodatkiem nadtlenku wodoru [11].
Autor: Lidia Koperwas
Literatura:
[1]. Murawa D., Banaszkiewicz T., Majewska E., Błaszczuk B., Sulima J., 2008. Zawartość azotanó (III) i (V) w wybranych gatunkach warzyw i ziemniakach dostępnych w handlu w Olsztynie w latach 2003-2004. BROMAT. CHEM. TOKSYKOL. – XLI, 2008, 1, str. 67–71. http://www.ptfarm.pl/pub/File/wydawnictwa/b_2008/1_2008/Artykul%2010%20Bromatologia%201-2008.pdf
[2]. Grudziński I.P., 1998. Effect of nitrates and nitrites of small intestine. ROCZN.PZH. 1998, 49, 321-330. http://www.google.pl/books?hl=pl&lr=&id=UWLSBa8tPYkC&oi=fnd&pg=PA321&dq=azotany+i+azotyny+w+%C5%BCywno%C5%9Bci&ots=Cxr8zRwMqF&sig=i2um3jEvClKxxWcacZcSxlhkdUo&redir_esc=y#v=onepage&q=azotany%20i%20azotyny%20w%20%C5%BCywno%C5%9Bci&f=false
[3]. Śmiechowska M., Przybyłowski P., 1999. Zawartość azotanów (V) i (III) w racjach pokarmowych studentów wyższej szkoły morskiej w Gdyni. ROCZ.PZH, 1999,NR 4, 385-390. http://books.google.pl/books?id=mHlhSpqkIi4C&pg=PA390&lpg=PA390&dq=azotany+i+azotyny+w+%C5%BCywno%C5%9Bci&source=bl&ots=w45k8jfwFX&sig=ujKNBaixUZDcPnTHnIh7XBrpI74&hl=pl&sa=X&ei=b0lfVPzxJsXsaMjOgcgF&ved=0CEgQ6AEwBzgK#v=onepage&q=azotany%20i%20azotyny%20w%20%C5%BCywno%C5%9Bci&f=false
[4]. Biegańska-Marecik R., Walkowiak-Tomczak D., Radziejewska-Bubzdela E., 2008. Zmiany zawartości azotanó (V) i (III) w szpinaku mało przetworzonym, pakowanym i przechowywanym w atmosferze modyfikowanej. ŻYWNOŚĆ. Nauka. Technologia. Jakość, 2008, 4 (59), 251 – 260. http://www.pttz.org/zyw/wyd/czas/2008,%204%2859%29/29_Bieganska.pdf
[5]. Gierak A., Leboda R., 1999. Analiza azotanów i bromianów powstających podczas dezynfekcji wody ozonem. Ochrona Środowiska, (4)75, 1999. http://www.os.not.pl/docs/czasopismo/1999/Gierak_4-1999.pdf
[6]. Sokołowska M., Włodek L., 2001. Dobre i złe strony tlenku azotu. Artykuł poglądowy. Folia Cardiol. 2001, tom 8, nr 5, 467–477 Copyright © 2001 Via Medica ISSN 1507–4145. http://czasopisma.viamedica.pl/fc/article/viewFile/24687/19754
[7]. Jaiswal M., LaRusso N.F.,Gores G.J., 2001. Nitric oxide in gastrointestinal epithelial cell carcinogenesis: linking inflammation to oncogenesis. American Journal of Physiology, Gastrointestinal and Liver Physiology. http://ajpgi.physiology.org/content/281/3/G626
[8]. http://www.wbns.uksw.edu.pl/sites/default/files/Cw_7_oznaczanie_anionow.pdf
[9]. Kolorymetryczne oznaczanie azotanów, fluorków i fosforanów. Instrukcja do ćwiczeń opracowana w Katedrze Chemii Środowiska Uniwersytetu Łódzkiego. http://www.chemia.uni.lodz.pl/kchs/index_pliki/Dokumenty/4.pdf
[10]. http://www.perfectwater.com.pl/instrukcje/Testoval%20AZOTANY%20instrukcja.pdf
[11]. Garbuliński T., 1955. Wykrywanie śladowych azotynów i azotanów przy użyciu fenoloftaleiny, nadtlenku wodoru i kwasu siarkowego. Notatki laboratoryjne. Roczniki Chemii 29, 1109 (1955). http://www.tadeusz-garbulinski.wroclaw.pl/files/002-55.pdf
[12].http://cctw.gig.eu/cscore/files/aparatura_cctw/wyposazenie_SC/Przebudowa%20zestawu%20analizatorow%20FIA_SC_opis.pdf
Recenzje