



- Biochemia
- Biofizyka
- Biologia
- Biologia molekularna
- Biotechnologia
- Chemia
- Chemia analityczna
- Chemia nieorganiczna
- Chemia fizyczna
- Chemia organiczna
- Diagnostyka medyczna
- Ekologia
- Farmakologia
- Fizyka
- Inżynieria środowiskowa
- Medycyna
- Mikrobiologia
- Technologia chemiczna
- Zarządzanie projektami
- Badania kliniczne i przedkliniczne



Elektroforeza kometkowa (Comet assay): przygotowanie wybranych etapów metody cz.II
 Elektroforezę kometkową charakteryzuje prostota wykonania, a także fakt, że z jej wykorzystaniem otrzymuje się powtarzalne wyniki badań. Z racji swego szerokiego zastosowania, comet assay należy do metod nieustannie modyfikowanych i ulepszanych. Tak więc, doczekała się już modyfikacji z wykorzystaniem przeciwciał albo hybrydyzacji z sondami molekularnymi (FISH), dzięki czemu możliwe jest szybkie i wybiórcze określenie zmian w obrębie DNA jądrowego [4].
Elektroforezę kometkową charakteryzuje prostota wykonania, a także fakt, że z jej wykorzystaniem otrzymuje się powtarzalne wyniki badań. Z racji swego szerokiego zastosowania, comet assay należy do metod nieustannie modyfikowanych i ulepszanych. Tak więc, doczekała się już modyfikacji z wykorzystaniem przeciwciał albo hybrydyzacji z sondami molekularnymi (FISH), dzięki czemu możliwe jest szybkie i wybiórcze określenie zmian w obrębie DNA jądrowego [4].
Metoda pozwala na analizę róznych typów uszkodzeń DNA, a także na badanie ich naprawy w indywidualnych komórkach. W 1978 roku Rydberg i Johanson, w trakcie prowadzonych badań wykazali, że jądra komórkowe poddawane lizie w warunkach alkalicznych pozostają stosunkowo skondensowanymi strukturami. Z kolei, po napromieniowaniu promieniowaniem jonizującym dochodzi dochodzi do rozluźnienia struktury DNA (co zależy od dawki promieniowania). Podstawą opisywanej metody stało się więc obserwowanie komórek po potraktowaniu ich 2M roztworem NaCl oraz detergentami anionowymi, które to prowadziły do powstawania „nukleoidów” zbudowanych z pętli DNA. Kilka lat później Ӧstling i Johanson (1984) wprowadzili etap krótkiego rozdziału elektroforetycznego komórek upzrednio poddanych lizie, a następnie zatopionych w agarozie na szkiełku mikroskopowym. Dalej, Singh i wsp. Zmodyfikowali całą procedurę metody i wprowadzili bufor alkaliczny do elektroforezy, a także dodatkowe rozwijanie DNA prowadzone bezpośrednio w alkalicznym roztworze elektroforetycznym. Zmiany te pozwalają na jednoczesną analizę pęknięć –jedno i –dwuniciowych, a także stanowią podstawę stosowanego testu kometkowego [8].
Choć metoda kometkowa (comet assay) jest stosunkowo prosta i szybka w wykonaniu, wymaga przeprowadzenia kilku kluczowych etapów, z których praktycznie każdy wymaga zastosowania innych buforów. W danych literaturowych pojawiają się rożne odmiany tego samego buforu (modyfikacje), które to powstają w trakcie wprowadzania zmian w pierwotnej metodologii. Jak już wspomniano, test kometkowy jest tak naprawdę modyfikowany od chwili wynalezienia jego pierwotnej wersji w 1978 roku (Rydberg i Johanson).
Bufory stosowane w metodzie kometkowej:
- Bufor lizujący (A): 2,5 M NaCL, 100 mM EDTA, 10 mM Trizma-base/Tris: Z podanych składników należy przygotowa bufor lizujący, dodając je do 800 ml wody destylowanej. Całość wymieszać dodając porcjami (ok. 7 g/L) NaOH. Po rozpuszczeniu wszystkich składników ustala sie pH, które powinno wynosić 10.0 . W tym celu stosuje się NaOH lub HCL. Po ustaleniu pH, próbkę uzupełnić wodą do objętości 900 ml i ponownie sprawdzić pH (doprowadzić do pH=10). Tak przygotowany bufor może być przechowywany w temperaturze pokojowej [1], [2].
- Bufor lizujący (B): W celu przygotowania tego buforu należy sporządzić 100ml 10% wodnego roztworu Triton X-100 tj. 90 ml wody destylowanej uzupełnić do 100 ml Tritonem X-100. Otrzymany bufor przechowuje się w temperaturze pokojowej.
- Pełny bufor lizujący (zmieszać bufor lizujący A z buforem lizującym B w stosunku: 90 ml :10 ml)- bufor schłodzić w lodówce przez 0,5 godziny. Pełny bufor lizujący należy przygotowywać bezpośrednio przed wykonaniem comet assay. Co ważne, uzupełnienie tego buforu 10% DMSO (dimetylosulfotlenkiem) chroni przed wtórnymi uszkodzeniami przez wolne rodniki, które uwalniane są z komórek w trakcie lizy [1], [2].
- Bufor PBS (roztwór soli fizjologicznej)
Ten wodny roztwór w składzie zawiera chlorek sodu oraz fosforan sodu, a dodatkowo może mieć także chlorek potasu oraz fosforan potasu. Głównym zadaniem PBS jest utrzymywanie stałe pH.Co więcej, stężenie jonów i ciśnienie osmotyczne buforu porównywalne jest do tego, jakie panuje w ludzkich płynach ustrojowych. Ze względu na dwie ważne cechy tj. izotoniczny charakter i brak toksyczność względem żywych komórek PBS powszechnie wykorzystywany jest w wielu róznych analizach [3].
Przygotowanie buforu PBS (w objętości 1 l):
W celu przygotowania roztworu PBS należy w 800 ml wody destylowanej kolejno rozpuścić:
- 80g NaCl (końcowa koncentracja (1X) równa 1,37M)
- 2 g KCl (27mM)
- 11,5 g Na2HPO4*7H2O (43mM)
- 1,9 g KH2PO4 (14 mM).
Po zmieszaniu wszystkich składników ustala się pH roztworu do 7.4, używając roztworu kwasu solnego lub wodorotlenku sodu. Po ustaleniu pH bufor uzupełnić wodą destylowaną do objętości 1 litra. Przed użyciem należy poddać bufor autoklawowaniu (sterylizacji) [3].
- Bufor alkaliczny (50 ml): NaOH (0,6g), 200 mM EDTA (250 µl), woda destylowana (49,75 ml). Całość mieszać aż do momentu całkowitego rozpuszczenia składników. Ze względu na obecność NaOH roztwór będzie się ogrzewał w trakcie przygotowywania. Po przygotowaniu buforu, pozostawić go do ostygnięcia [7].
- Bufor elektroforetyczny (1xTBE): W celu przygotowania 10x stężonego buforu TBE należy zmieszać: Tris (108g), kwas borowy (55g), EDTA (9,3g). Składniki rozpuścić w 900 ml wody destylowanej. Bufor przechowywać w temperaturze pokojowej [7].
- Bufor zapobiegający blaknięciu (wykorzystywany w przypadku, gdy występuje zanik próbek)
W 50 ml probówce należy zmieszać: dichlorowodorek p-fenylenodiaminę (500 mg), 1x PBS (4,5 ml). Następnie, dodać około 400 µl 10 M NaOH, wkraplając do momentu aż pH roztworu osiągnie 7.5 – 8.0. Dodać 1x PBS, aby zwiększyć objętość do 5 ml i 45 ml glicerolu- do końcowej objętości równej 50 ml. Całość przygotowanej próbki dokładnie zvorteksować,a w razie konieczności nanosić w objętości 10 µl na próbkę. Przygotowany bufor można przechowywać do jednego miesiąca w temperaturze -20°C, a w trakcie przechowywania może wystąpić ciemnienie roztworu [7].
Przygotowanie fragmentów tkanek do analizy
Mały kawałek tkanki, która ma zostać poddana analizie w teście kometkowym, należy umieścić w 1-2 ml lodowatego 1x stężonego buforu PBS (wolnego od jonów Mg2+ i Ca2+) z dodatkiem 20 mM EDTA. Za pomocą skalpela pociąć tkankę na bardzo małe fragmenty, po czym pozostawić na 5 minut. Na tym etapie liczy się komórki, wiruje i ponownie rozpuszcza otrzymaną zawiesinę do 105 komórek/ml w lodowatym (wolnym od jonów magnezu i wapnia) PBS (1x). Następnie, do próbki dodać 1-2 ml lodowatego 20 mM roztworu EDTA w 1x PBS, tkankę zmielić na bardzo małe kawałki i pozostawić do odstania na 5 minut. Odzyskanie zawiesiny komórek pozwoli na uniknięcie przenoszenia zanieczyszczeń. Na tym etapie ponownie zlicza się komórki i zawiesza ich osad w 1xPBS (jak wyżej) [7].
Przygotowanie preparatów do comet assay [1],[2].
- 0,5% agarozę o niskiej temperaturze topnienia przygotowanej na buforze PBS, należy rozpuścić we wrzącej łażni wodnej. Gdy agaroza się rozpuści, przenieść ją w objętości 150 µl do probówek typu eppendorf. Tak przygotowane próbki umieścić w łaźni wodnej o temperaturze 42°C (pozostawiając do ustalenia temperatury)
- Analizowane komórki, należy zebrać z hodowli i policzyć w hemocytometrze. Odpowiednią ilość komórek pzrenieść do przygotowanych wcześniej probówek eppendorf , po czym osadzić je przez wirowanie (300x g, 5 minut w temperaturze pokojowej).
- Po wirowaniu, należy zebrać supernatant, a osad rozbić i zawiesić w 1 ml buforu PBS, próbki ponownie zwirować (jak wyżej), usunąć powstały supernatant, a osad zawiesić ponownie w buforze PBS do ok. 10000 komórek/10 µl (tj. 106 komórek/ ml).
- 20 µl zawiesiny komórek połączyć ze 150 uL 0,5% LMP agarozy, całość wymieszać, po czym nanieść na szkiełko podstawowe pokryte uprzednio 30 µl agarozy NMP (1% agaroza o niskiej temperaturze topnienia). Tak przygotowany preparat należy nakryć szkiełkiem nakrywkowym (lekko dociskając) i pozostawić w lodówce na ok 10 minut.
- Zsunąć (nie unosić) szkiełko nakrywkowe (ewentualnie nanieść na preparat trzecią warstwę agarozy LMP), szkiełko zanurzyć w świeżym, zimnym buforze do lizy (bufor A + bufor B), inkubować w buforze przez 1 godzinę w 4°C (lub przez noc).
- Po upływie czasu inkubacji, zebrać bufor lizujący, a następnie szkiełka zalać zimną wodą destylowaną (szkiełka delikatnie zamieszać, po czym odmyć z nich bufor lizujący). Czynność tę należy powtórzyć 3-krotnie (inkubacja w świeżej porcji wody destylowanej powinna wynosić 5 minut).
- Na kolejnym etapie szkiełka należy zalać świeżym i zimnym buforem do rozdziału elektroforetycznego o pH>13. Inkubować je w buforze przez 15 minut (4°C), zaś po upływie czasu inkubacji zebrać bufor. Czynność powtarzać do momentu, aż całkowity czas inkubacji w buforze elektroforetycznym będzie trwać minimum 30 minut.
- Tak przygotowane szkiełka przenosi się do aparatu elektroforetycznego , uprzednio wypełnionego świeżym, zimnym buforem elektroforetycznym. Rozdział elektroforetyczny prowadzić przy napięciu ok 75 V/cm (natężenie do 300mA), przez 30 minut w temperaturze 4°C.
- Po rozdziale, szkiełka przenieść do czystego naczynia, gdzie przemyć je (3x) zimną wodą destylowaną. Następnie, zalewa się je buforem neutralizacyjnym, w którym inkubuje się przez 10 minut. Po inkubacji szkiełka ponownie przemywa się wodą destylowaną (3x).
- Końcowym etapem przygotowania preparatów jest ich zanurzenie w 70% roztworze etanolu (5-minutowa inkubacja). Tak przygotowane szkiełka suszy się w temperaturze pokojowej lub cieplarce (37°C) i barwi [1],[2].
Olive P.L. i Banáth J.P.(2006), przedstawili wykorzystanie metody kometkowej do pomiaru uszkodzenia DNA w poszczególnych komórkach eukariotycznych. Choć większość badań opiera się na wykorzystaniu metody kometkowej do pomiaru przerw w pojedynczej nici DNA, wprowadzenie odpowiednich modyfikacji w metodzie umożliwia wykrywanie przerw również w podwójnej nici DNA, połączeń krzyżowych czy jąder apoptotycznych. Czułość testu w przeprowadzonym badaniu wynosiła około 50 pęknięć nici DNA na diploidalną komórkę ssaków. Ponadto, metoda ta znalazła także zastosowanie do komórek drożdży, pierwotniaków czy roślin. Przedstawiony przez Olive P.L. i Banáth J.P.(2006) protokół może być zakończony w mniej niż 24 godziny, co potwierdza,że comet assay jest stosunkowo szybką metodą analizy [5].
Analiza komórek apoptotycznych
W trakcie procedu apoptozy dochodzi do uszkodzenia (degradacji) komórkowego DNA. Degradacja zachodzi pod wpływem enzymu endonukleazy, która to aktywowana jest przez kaspazy. W trakcie tego procesu DNA hydrolizowane jest preferencyjnie w miejscach pomiędzy nukleosomami. Na skutek degradacji DNA rozpada się na fragmenty o długości ok 180 par zasad bądź ich wielokrotności (tj. 360pz, 560pz itd). W pzreprowadzonych badaniach wykazano, że ilość powstających fragmentów DNA zależna jest od tego, w jakich zachodzi komórkach, tj. czy są to komórki apoptotyczne czy nekrotyczne. Fragmentacja dla komórek apoptotycznych jest wyższa niż dla komórek nekrotycznych, na czym też test kometkowy się opiera. Comet assay umożliwia odróżnienie dwóch typów śmierci komórek (apoptotycznej i nekrotycznej) właśnie na tej podstawie [6].
Dla komórek nekrotycznych i apoptotycznych wykonuje się w identyczny sposób jak dla wszystkich innych typów komórek. Naniesione i utrwalone na szkiełku komórki poddaje się rozdziałowi elektroforetycznemu, podczas którego (po przyłożeniu napięcia pofragmentowane DNA opuszcza komórkę i migruje w żelu w kierunku anody). Po zakończeniu rozdziału elektroforetycznego, DNA wybarwia się w celu jego uwidocznienia . Najczęściej stosuje się w tym celu bromek etydyny. DNA, które opuściło komórkę po uwidocznieniu tworzy charakterystyczny wzór- podobny do ogona komety, zaś to pozostałe w komórce wygląda jak głowa komety. W przypadku, gdy nie nie doszło do pęknięć lub cięcia DNA, obserwuje się świecenie tylko w obrębie komórki tj. głowy komety. Wszystko wynika z faktu, że nie pocięte DNA jest zbyt duże, by pod wpływem pola elektrycznego opuścić komórkę, w związku z czym pozostaje uwięzione w strukturach komórkowych. Komórki nekrotyczne również dają fluoryzujący ogon, aczkolwiek precyzyjny pomiar parametrów, które dokładnie opisują intensywność świecenia ogona pozwala na ich odróżnienie od komórek apoptotycznych [6].
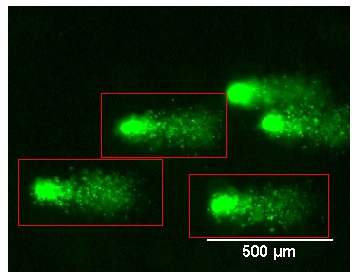
Zdjęcie: Kometki, http://library.wolfram.com/examples/cometassay/
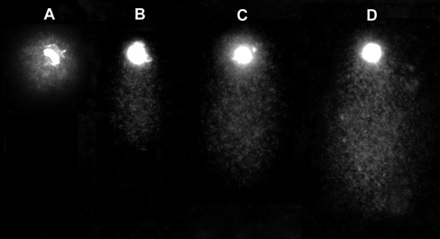
Zdjęcie: Komórki HeLa po analizie comet assay, http://jhc.sagepub.com/content/59/7/655/F2.expansion.html
Wojewódzka M. i wsp. (2000) przeprowadzili test kometkowy na komórkach krwi (limfocytach) pobranych z żyły. W badaniach wykorzystano znacznie skróconą metologię (w porónaniu do opisywanych w literaturze procedurami), dzięki czemu uproszczono i znacznie skrócono poszczególne etapy testu. W trakcie doświadczenia uzyskano dobrą korelację pomiędzy wielkością uszkodzenia (mierzoną mierzoną tzw. „momentem ogonowym”), a zastosowaną do badań dawką promieniowania jonizującego. Pomimo wprowadzonych przez badaczy modyfikacji w metodzie dowiedzono, że kinetyk anaprawy podwójnoniciowych pęknięć DNA jest jak najbardziej zgodna z podawanymi danymi literaturowymi [8].
Liza komórek krwi (limfocytów) do testu kometkowego (procedura wg Wojewódzka M. i wsp., 2000):
Przed rozpoczęciem tego etapu przygotowano podstawowy bufor do lizy (tj.: 2,5 M NaCl, 100mM EDTA, 10 mM Tris-HCl, 1% sarkozyl-N-laurylosarkozyna, bufor o pH=9.5). Bezpośrednio przed użyciem do buforu dodano 0,5% Triton X-100 oraz 10% DMSO (dimetylosulfotlenek). Całość mieszano z wykorzystaniem mieszadłą magnetycznego (około 20 minut) i schładzano w lodówce. Do tak przygotowanego zimnego buforu wkładano szkiełka z naniesionymi na nie komórkami zawieszonymi w agarozie. Etap lizy prowadzono w ciemności przez 1-2 godzin w temperaturze 4°C.
Płukanie preparatów po lizie (procedura wg Wojewódzka M. i wsp., 2000):
Preparaty przemywano 3-krotnie z wykorzystaniem buforu do elektroforezy (tj.: 300 mM octan sodowy, 100 mM Tris-HCl o pH=8.5). Preparaty pozostawiono w świeżej porcji buforu na co najmniej 1 godzinę, w celu dokładnego wypłukania resztek soli (pozostałych po etapie lizy).
Rozdział elektroforetyczny (procedura wg Wojewódzka M. i wsp., 2000):
W aparacie do elektroforezy umieszczono wypłukane szkiełka podstawowe, a rozdział prowadzono przy napięciu 14V i natężeniu 11-12 mM, przez 1 godzinę w temperaturze nie przekraczającej 10°C. Cały rozdział prowadzono w ciemności. Po skończonym rozdziale szkiełka zostały 3 razy przepłukane w w buforze neutralizującym (tj.: 0,4 M Tris o pH= 7.5). Szkiełka następnie osuszono z nadmiaru roztworu i poddano barwieniu (1µM roztworem DAPI), po czym nakryto preparat szkiełkiem nakrywkowym, po czym preparaty włożono do pudełka wyłożonego wilgotną bibułą i przechowywano w lodówce przez około 20 godzin. Przygotowane w ten sposób preparaty mikroskopowe poddawano następnie analizie komputerowej[8].
Wprowadzone przez Wojewódzką M. i wsp. (2000) modyfikacje w metodzie kometkowej, polegające na prowadzeniu testu w pH obojętnym, pozwoliły na otrzymanie komet o bardziej zwartym zarysie, niż w testach wykonywanych w publikacjach innych autorów. Wcześniej opisane procedury (przed modyfikacją powyższych autorów) dostarczały „obrazy” komet o bardzo wydłużónych i rozmytych ogonach, co obniżało precyzyjnośc pomiarów, a momentami wręcz uniemożliwiało ich przeprowadzenie [8].
Analiza komet metodą Gedika
Do oceny skali uszkodzenia materiału genetycznego po teście kometkowym zazwyczaj wykorzystuje się specjalistyczne oprogramowanie do komputerowej analizy obrazu. Innym, prostym sposobem pozwalającym na analize obrazu komet jest tzw. metoda Gedika i wsp. (1992), która opiera się na klasyfikacji uszkodzeń jądrowego DNA w komórce do pięciu klas.
Skala ta obejmuje charakterystyczne oznaczenia:
1) N –brak uszkodzeń (mniej niż 5% DNA w ogonie komety)
2) L-małe uszkodzenia (5- 25% DNA w ogonie komety)
3) M- średnie uszkodzenia (20-40% DNA w ogonie komety)
4) H- duże uszkodzenia (40 – 95% DNA w ogonie komety)
5) T-całkowite uszkodzenia (ponad 95% DNA w ogonie komety)[4].
Autor: Lidia Koperwas
Literatura:
[1]. http://www.kucharczyk.com.pl/instrukcje/poradykometa.pdf . Dr Marcin Schmidt, Uniwersytet Przyrodniczy w Poznaniu, http://www.up.poznan.pl/~mschmidt/
[2]. http://www.kucharczyk.com.pl/
[3]. http://www.e-biotechnologia.pl/Artykuly/sol-fizjologiczna
[4].Brzuzan P., Woźny M., Łuczyński M.K., 2007. Toksykologia molekularna, przewodnik do ćwiczeń, Olsztyn 2001. http://biotechnology.keyland.biz/pdf/tox_mol_updated.pdf
[5]. Olive P.L. i Banáth J.P., 2006. The comet assay: a method to measure DNA damage in individual cells. http://www.ncbi.nlm.nih.gov/pubmed/17406208 (abstract)
[6]. http://bioinfo.mol.uj.edu.pl/articles/Kacprzyk04
[7]. http://www.amsbio.com/protocols/Comet_Assay.pdf
[8]. Wojewódzka M., Grądzka I, Buraczewska I., 2000. Test kometowy w pH obojętnym. Instytut Chemii i Techniki Jądrowe, Warszawa 32/28. Raporty IchTJ.Seria B nr 2/2000. http://www.iaea.org/inis/collection/NCLCollectionStore/_Public/32/032/32032373.pdf
Tagi: test kometkowy, comet assay, DNA jądrowe, uszkodzenia DNA, bufory, liza komórek, analiza, komórka apoptotyczna, analiza kometek, test Gedika, rozdział elektroforetyczny
wstecz Podziel się ze znajomymi
Recenzje