



- Biochemia
- Biofizyka
- Biologia
- Biologia molekularna
- Biotechnologia
- Chemia
- Chemia analityczna
- Chemia nieorganiczna
- Chemia fizyczna
- Chemia organiczna
- Diagnostyka medyczna
- Ekologia
- Farmakologia
- Fizyka
- Inżynieria środowiskowa
- Medycyna
- Mikrobiologia
- Technologia chemiczna
- Zarządzanie projektami
- Badania kliniczne i przedkliniczne




Mechanizm procesów katalitycznych
Wprowadzenie
Katalizator zmienia szybkość reakcji chemicznej, przy czym nie zużywa się podczas jej przebiegu. Własność ta jest przeciwieństwem działania induktora, który również przyspiesza reakcję chemiczną ale w procesie indukcji jest zużywany. Przykładami induktorów są związki wytwarzające wolne rodniki, które z kolei inicjują reakcje łańcuchowe polimeryzacji, czy kopolimeryzacji monomerów organicznych. W efekcie tego działania otrzymywane są tworzywa sztuczne o bardzo szerokim zastosowaniu zarówno w przemyśle, jak i życiu codziennym.
Spośród stosowanych obecnie katalizatorów, główną rolę odgrywają katalizatory przyspieszające reakcje chemiczne, nazywane są one katalizatorami dodatnimi. Katalizatorami ujemnymi są inhibitory, które zmniejszają szybkość reakcji chemicznej. Efekt ich działania ze zwiększenia wartości energii aktywacji reakcji. Podobnie jak w przypadku katalizatorów dodatnich należy rozróżnić inhibitory od substancji, których działanie polega na wychwytywaniu (zmiataniu) reaktywnych indywiduów chemicznych, jakimi są rodniki, przez co reakcja zostaje zahamowana. Substancje te, podobnie jak induktory, zużywane są w toku reakcji. Stężenie katalizatora lub inhibitora, niezbędne do wyraźnej zmiany szybkości reakcji jest na ogół bardzo niewielkie, kilka do kilkunastu rzędów wielkości mniejsze niż stężenie reagentów.
Reakcja chemiczna, w której bierze udział katalizator nazywana jest reakcją lub procesem katalitycznym. Działanie katalizatora określa się mianem katalizy.
Katalizatory homogeniczne i heterogeniczne
Reakcja katalityczna może zachodzić w układzie jednofazowym lub wielofazowym. W zależności od tego mamy podział na katalizatory homogeniczne i heterogeniczne. W pierwszym
przypadku katalizator tworzy jedna fazę wspólnie z mieszaniną reakcyjną. Proces taki nazywany jest katalizą homogeniczną (jednofazową). Jeżeli proces katalityczny zachodzi na granicy faz pomiędzy powierzchnią katalizatora i reagentami, wówczas mamy do czynienia z katalizą heterogeniczną. Wszystkie znane katalizatory heterogeniczne są substancjami stałymi, często o bardzo rozbudowanej powierzchni właściwej. Katalizy heterogenicznej nie należy mylić z katalizą przeniesienia międzyfazowego. Zjawisko to zachodzi na granicy dwóch nie mieszających się ze sobą cieczy lub cieczy i nierozpuszczalnego w niej ciała stałego, jednakże katalizator jest w tym przypadku rozpuszczony w mieszaninie reakcyjnej, a jego funkcja polega na przenoszeniu cząsteczek reagentów przez granicę faz. Pomimo iż katalizatory heterogeniczne nie zmieniają w procesie katalitycznym swojego składu chemicznego, może mieć miejsce zmiana ich struktury fizycznej (zmiana formy krystalograficznej, pojawienie się defektów sieciowych, przejście w formę amorficzną.
Mechanizm procesu katalitycznego
Na wstępie należy zaznaczyć, iż mechanizm działania wielu katalizatorów nie został dotychczas do końca wyjaśniony, zaś katalizatory do poszczególnych reakcji chemicznych dobiera się często w sposób eksperymentalny, to i tak kataliza odgrywa bardzo ważną rolę w chemii. Większość procesów technologicznych stosowanych w przemyśle to właśnie reakcje katalityczne.
W przypadku katalizy heterogenicznej, pierwszym etapem procesu katalitycznego jest zjawisko adsorpcji fizycznej lub chemicznej reagentów na powierzchni katalizatora, który występują w postaci stałej. Przyjmuje się, że powierzchnia katalizatorów tego typu jest niejednorodna (porowata), a zjawisko katalizy ma miejsce na istniejących na niej centrach aktywnych. Pierwszym efektem adsorpcji substratów jest zwiększenie ich stężenia w bezpośrednim sąsiedztwie katalizatora, co przyczynia się do wzrostu szybkości reakcji. Dalszym efektem jest deformacja chmury elektronowej cząsteczek substratu, co prowadzi do zwiększenia ich aktywności. Czasem przyciąganie adsorpcyjne jest na tyle duże, że prowadzi do rozerwania wiązań w cząsteczce.
W przypadku chemisorpcji, cząsteczki reagentów tworzą wiązania z atomami katalizatora, w wyniku czego całkowitej zmianie ulega ich reaktywność. Wiązania w cząsteczkach substratów są rozrywane, zaś cząsteczki przechodzą w nowe, pośrednie indywidua, istniejące jako jednocząsteczkowa warstwa na powierzchni katalizatora. Następnie, w procesie desorpcji tworzą się nowe wiązania, prowadzące do tworzenia się cząsteczek produktów.
W katalizie homogenicznej katalizator reaguje z cząsteczkami jednego z substratów, tworząc produkt pośredni. Utworzony produkt pośredni reaguje z cząsteczką drugiego substratu, z odtworzeniem cząsteczki katalizatora i utworzeniem cząsteczki produktu. Proces ten powtarza się w sposób ciągły do wyczerpania substratu.
Mechanizm katalizy homogenicznej w reakcjach metatezy
Historia metatezy sięga początku lat 50-tych. W okresie tym trwały intensywne badania nad poznaniem katalitycznego działania metali przejściowych na układy zawierające węglowodory nienasycone. Zainteresowanie owym zagadnieniem wynikało bezpośrednio z eksperymentalnych wyników Zieglera i Natty oraz szybko rozwijających się metod polimeryzacji etylenu na katalizatorach heterogenicznych [1]. Ziegler i Natta zajmowali się wówczas katalitycznymi własnościami związków metaloorganicznych.
Na rok 1954 przypada pierwszy opisany przykład metatezy olefin (związki z wiązaniem podwójnym). Była to polimeryzacja norbornenu przeprowadzona w obecności TiCl4 (tetrachlorek tytanu) przez Andersona i Merckliego w laboratoriach firmy Du Pont [1].
W roku 1964 Banks i Bailey z Philips Petroleum opublikowali pierwsze sprawozdanie opisujące dysproporcjonowanie olefin acyklicznych na katalizatorze M(CO)6-Al2O3 (M = Mo - molibden lub W - wolfram). W wielu publikacjach można odnaleźć cytowania tej daty niejako określającej rok odkrycia metatezy [1,2]. W tym samym czasie grupa Natty w Montedison odkryła, iż cyklobuten i cyklopenten ulegają polimeryzacji z otwarciem pierścienia w obecności katalizatorów: MoCl5-AlEt3, TiCl4-AlEt3 i WCl6-AlEt3 [1].
Następnie w 1967 roku Calderon wraz ze swoim zespołem opisał niezwykle aktywny układ katalizujący dysproporcjonowanie 2-pentenu w temperaturze pokojowej. Katalizatorem tym był układ WCl6-EtAlCl2-EtOH w stosunku (1:4:1) [1]. Calderon też jako pierwszy sformułował termin „metateza”, jako wymiana atomów lub grup atomów pomiędzy dwiema cząsteczkami.
Herisson i Chauvin zaproponowali w 1971 roku karbenowy mechanizm metatezy. Mechanizm ten zakłada obecność w środowisku reakcji związków typu metalokarbenowego [M]=CM, które tworzą z olefiną pośredni produkt, a mianowicie metalocyklobutan, który następnie ulega rozpadowi z utworzeniem nowej olefiny (Rys. 1.).
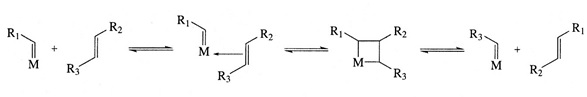
Rysunek 1. W pierwszym etapie oddziaływanie karbenu z olefiną ma prawdopodobnie charakter π-kompleksu (od lewej), później zaś powstaje związek cykliczny (środek). Po każdym cyklu następuje odtworzenie aktywnego kompleksu alkilidenowego, który może przyłączać kolejną cząsteczkę olefiny (po prawej).
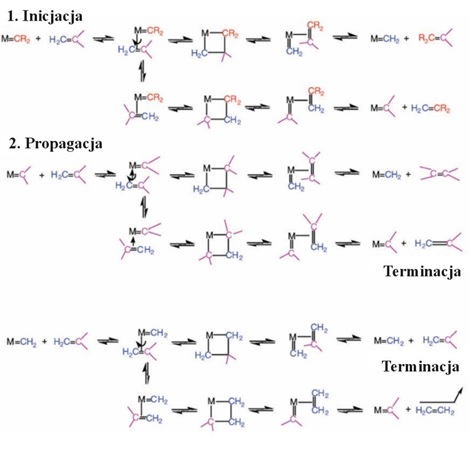
Rysunek 2. Karbenowy mechanizm metatezy zaproponowany przez Herissona i Chauvina uwzględniający etap inicjacji, propagacji oraz terminacji reakcji [2].
W roku 1974 Schrock wyizolował kompleks metalo-alkilidenowy [Ta=CHBut(CH2But)3] (Ta - tal) [2]. Sześć lat później jako pierwszy otrzymał jednocząsteczkowy kompleks katalizujący metatezę [Lata=CHBut]. Znaczenie metatezy zaczęło nabierać coraz większego znaczenia w syntezie organicznej. Metoda ta ograniczała się jednak do prac laboratoryjnych w małej skali.
W roku 1992 Grubbs odkrył trwały na powietrzu karbenowy kompleks rutenu promujący metatezę [Ru=C=CHPh(PR3)2Cl2], zaś trzy lata później zaowocowało to otrzymaniem przez Grubbsa katalizatora umożliwiającego prowadzenie metatezy olefin w warunkach przemysłowych [Ru=CHPh(PCy3)2Cl2] [2].Od tamtej chwili katalizatory metatezy zaczęto produkować i stosować na dużą skalę. Do dziś powstało wiele kompleksów promujących reakcje metatezy olefin należących do rozmaitych klas związków (Rys. 3).
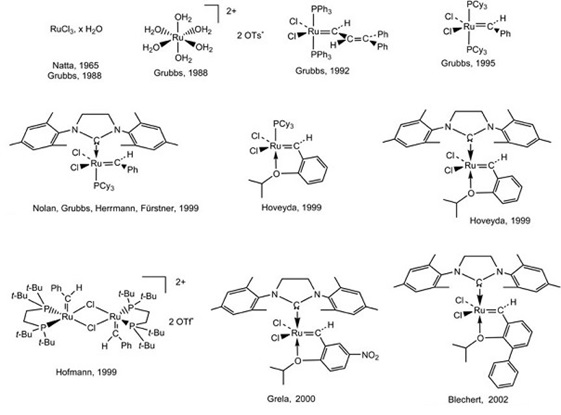
Rysunek 3. Kierunki rozwoju katalizatorów rutenowych, daty odkrycia [2].
Katalizatory najczęściej stosowane w reakcjach metatezy
Katalizatory niezdefiniowane
Najstarsze historycznie substancje promujące reakcje metatezy zostały nazwane katalizatorami niezdefiniowanymi. Nazwa ta dotyczyła ich struktury. Ze względu na niezdefiniowaną do końca budowę trudno było przewidywać mechanizm oraz kierunkowość ich działania. Dlatego też nie znalazły one szerokiego zastosowania [1]. Przełomem okazały się natomiast katalizatory o ściśle określonej strukturze. To właśnie one zapoczątkowały nagły rozwój i zainteresowanie metatezą jako jedną z nowoczesnych metod syntezy.
Katalizatory zdefiniowane
W obecnej chwili szeroko stosowane są katalizatory o ściśle zdefiniowanej strukturze. Metateza wymaga bowiem dobrego przewidywania kierunkowości reakcji. Gwarantem tego jest znajomość struktury katalizatora, która w połączeniu z optymalnymi warunkami narzuca formę otrzymywanego produktu.
Katalizatory Molibdenowe i Wolframowe Schrocka
Należące do starszej generacji katalizatorów są one jednak niezwykle skuteczne w reakcjach metatezy z zamknięciem pierścienia prowadzących do utworzenia wiązania podwójnego trój- lub nawet czteropodstawionego. Ogromną zaletą tych związków jest również to, iż dobrze reagują z olefinami ubogimi elektronowo. Same są mało wrażliwe na własności elektronowe olefin.
Jednakże katalizatory Schrocka wykazują wysoką wrażliwość na tlen. Reakcję zatem należy prowadzić w środowisku gazu obojętnego z wykorzystaniem techniki Schlenka.
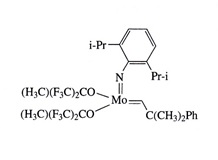
Rysunek 4. Alkilidenowy kompleks molibdenu. Katalizator Schrocka [3].
Dużą zaletą tych kompleksów jest możliwość łatwego wprowadzania ligandów zawierających fragmenty chiralne w sferę koordynacyjną metalu, co pozwala prowadzić reakcję asymetryczną [3].
Katalizator Grubbsa pierwszej generacji
Następnym katalizatorem najczęściej używanym w reakcjach metatezy jest karbenowy kompleks rutenu odkryty w 1995 roku przez Grubbsa 1 (Rys. 3). Katalizator ten znalazł największą liczbę zastosowań w syntezie organicznej ze względu na łatwość przechowywania, względną stabilność na powietrzu i wobec wilgoci oraz dużą tolerancję względem grup funkcyjnych.
Jego podstawową wadą jest niska skuteczność w reakcjach metatezy z udziałem reagentów zawierających podstawione wiązania podwójne oraz zawierających grupę elektronoakceptorową przy wiązaniu podwójnym jak np. α,β-nienasycone związki karbonylowe.
Ciekawostką jest to, że reakcje z udziałem katalizatora Grubbsa można prowadzić w bardzo polarnych ośrodkach jak woda lub metanol. W przypadku tego kompleksu stosowane są również ośrodki dwufazowe.
Katalizatory rutenowe drugiej generacji
Katalizatory zaliczane do tej grupy są rutynowymi kompleksami posiadającymi zamiast jednego ligandu fosfinowego cząsteczkę heterocyklicznego karbenu (ang. N-heterocyclic carbene, NHC). Ligand ten może zawierać w pierścieniu heterocyklicznym wiązanie nienasycone(tzw. Katalizator Nolana) lub nasycone (katalizator Grubbsa drugiej generacji) (Rys. 3). Kompleksy charakteryzują się znaczną trwałością podczas przechowywania, względną odpornością na powietrze i wilgoć oraz wysoką tolerancją względem rozmaitych grup funkcyjnych.
Przeprowadzenie reakcji metatezy z udziałem tych katalizatorów jest proste, zaś powstające produkty zawierają wiązania podwójne trój-, a nawet czteropodstawione. Warto dodać, iż są one również skuteczne w reakcjach olefin zawierających grupę elektronoakceptorową przy wiązaniu podwójnym.
Katalizator Hoveydy pierwszej generacji
To odkryty przez Hoveydę w 1999 roku rutenowy karben, w którym metal chelatowany jest tlenem z grupy 2-izopropoksybenzylidenowej (Rys. 3). Kompleks ten okazał się niezwykle aktywnym katalizatorem metatezy olefin. W porównaniu z wcześniej wymienionymi kompleksami wykazuje on doskonałą tolerancję na powietrze i wilgoć. Można go zatem stosować do różnych reakcji metatezy bez konieczności zachowania ekstremalnie bezwodnych i beztlenowych warunków. Katalizator Hoveydy ma zdolność do samoregeneracji po wyczerpaniu się substratu. W niektórych przypadkach można go odzyskać za pomocą chromatografii kolumnowej z bardzo wysoką wydajnością.
Katalizator Hoveydy drugiej generacji
Jest zmodyfikowanym katalizatorem Hoveydy pierwszej generacji, w których ligand fosfinowy zastąpiono nasyconym ligandem NHC [4].Kompleks ten wykazuje wysoką skuteczność w reakcjach metatezy z zamknięciem pierścienia dwu- i trójpodstawonych olefin. Znalazł również zastosowanie w reakcjach otwarcia pierścienia oraz metatezy krzyżowej.Po zakończeniu reakcji komplek tego typu można wydzielić na kolumnie chromatograficznej i ponownie użyć do reakcji. Trafne jest zatem określenie go jako samoodnawiającego się w procesie metatezy (Rys. 3).
Mechanizm katalizy heterogenicznej w reakcjach radioznacznikowania peptydów izotopami jodu
Molekuły biologicznie czynne naturalnie występują w bardzo niskich stężeniach, których nie można analizować tradycyjnymi metodami. Wprowadzenie radioizotopu do struktury białka pozwala obniżyć próg detekcji przy zastosowaniu detektorów promieniowania β- lub γ. Radioznacznikowanie izotopami jodu jest techniką polegającą na włączeniu w strukturę peptydu izotopów 123I, 125I lub 131I, przede wszystkim w celu przeprowadzenia badań in vitro, ale izotopy te mogą też być wprowadzone w celach terapeutycznych lub diagnostycznych.
Obecnie istnieją trzy podstawowe metody jodowania, które są powszechnie wykorzystywane do znakowania biomolekuł używanych w terapii i diagnostyce medycznej. Mowa tu o użyciu ampuł Iodo-Gen inkorporowanych po wewnętrznej stronie katalizatorem heterogenicznym [5], metodzie wykorzystującej jako katalizatora chloraminę-T [6] oraz metodzie z wykorzystaniem katalitycznych właściwości laktoperoksydazy [7].
W zależności od stosunku molowego użytego jodu do grup tyrozynowych i histydynowych występujących w strukturze peptydu oraz od wydajności katalizatora, mogą powstawać pochodne monojodowe oraz pochodne jodowe wyższych rzędów.
Wprowadzenie jodu do struktury białka lub peptydu z reguły nie wpływa na jego właściwości ze względu na niewielkie rozmiary atomu w porównaniu do całej biomolekuły.
Zastosowanie techniki Iodo-Gen, jest najczęstszym sposobem znakowania białek izotopami jodu (Rys. 5).

Rysunek 5. Ampułki Iodo-Gen oraz immobilizowany na wewnętrznej powierzchni ampułki katalizator.
Występujący tu katalizator w sposób trwały związany jest z powierzchnia szkła borokrzemowego, dzięki czemu nie miesza się z roztworem reagentów i w łatwy sposób może zostać oddzielony od środowiska reakcji. Odbywa się to poprzez przelanie mieszaniny reakcyjnej do innej ampułki. Mechanizm działania tego katalizatora polega na wytworzeniu produktu przejściowego z anionem jodkowym, a następnie przekształceniu go w kation i substytucję elektrofilową takiego kationu do pierścienia aromatycznego substratu. W wyniku tej reakcji powstaje podstawiony jodem produkt, zaś sam katalizator ulega odtworzeniu. Jest to klasyczny przykład katalizy heterogenicznej.
Podsumowanie
Techniki z zastosowaniem substancji chemicznych, które zmieniając szybkość reakcji, przy czym same nie ulega zużyciu podczas jej przebiegu są bardzo szeroko rozprzestrzenione. Nierozłącznie jest to związane z rozwojem naszej cywilizacji. Postęp technologiczny nastawiony jest na pozyskiwanie nowych produktów najszybszą, najbardziej efektywną oraz przyjazną środowisku drogą. Katalizatory reakcji chemicznych spełniają wszystkie te wymogi, gdyż działają wielce efektywnie przy bardzo niskich stężeniach, a przy tym można je odzyskiwać, regenerować i stosować wielokrotnie. W dzisiejszych czasach opracowywane są nowe i bardzo specyficzne katalizatory reakcji. Przytoczone powyżej przykłady odbiegają od klasycznego pojęcia katalizy, a mimo wszystko stosowane są na skalę przemysłową. Dzięki katalizatorom rutenowym możliwa jest polimeryzacja oraz kopolimeryzacja olefin o bardzo specyficznym działaniu i zastosowaniach medycznych. Natomiast dzięki katalitycznemu radioznacznikowaniu białek możliwe jest obrazowanie SPECT oraz terapia antynowotworowa na przykład raka tarczycy czy piersi. Prócz przytoczonych wcześniej zastosowań nie można zapomnieć o otrzymywanych na ogromna dziś skalę tworzyw sztucznych, które towarzysza nam na co dzień w rozmaitej formie. Mowa tu chociażby o sprzęcie AGD, ubraniach, elektronice, opakowaniach plastikowych, czy też samochodach. Bardzo ważny jest aspekt końcowego zagospodarowania produktów otrzymywanych przez katalizę, a mianowicie biodegradowalności.
To bardzo szerokie zagadnienie jest jednak odrębnym tematem.
Autor: Karolina Wójciuk
Literatura:
1. J. Handzik, J. Ogonowski, 1998. Metateza olefin, Kraków i literatura tam cytowana.
2. Astruc, D., 2005. New J. Chem., 29, 42-56 i zestawienia tam publikowane.
3. Kingsbury, J. S.; Harrity, J. P. A.; Bonitatebus, P.J.; Hoveyda, A. H., 1999. J. Am. Chem. Soc., 121, 791-7994.
4. Garber, S. B.; Kingsbury, J. S.; Gray, B. L.; Hoveyda, A. H., 2000. J. Am. Chem. Soc., 122, 8168-8176.
5. Fraker, P.J., Speck, J.C., 1978. Protein and cell membrane iodinations with a sparingly soluble chloramide, 1,3,4,6-tetrachloro-3a,6a-diphenylglycoluril. Biochem. Biophys. Res. Commun. 80(4), 849-857.
6. Wright, P.J., di Mayorca, G., 1976. Radioisotopic Labeling of Human Papovavirus (BK) by Iodination and Reductive Alkylation. J. Virol. 19(2), 750-755.
7. Marchalonis, J.J., 1969. An Enzymic Method for the Trace lodination of Immunoglobulins and other Proteins. Biochem. J. 113(2), 299-305.
Tagi: katalizator, kataliza, reakcja katalityczna, katalizator heterogeniczny, katalizator homogeniczny, produkt przejściowy, mechanizm katalizy, metateza, radioznacznikowanie, powierzchnia katalizatora, immobilizowany, energia aktywacji, energia kinetyczna
wstecz Podziel się ze znajomymi
Recenzje